

Assessing protein loop flexibility by hierarchical Monte Carlo sampling
- PMID: 21743800
- PMCID: PMC3129859
- DOI: 10.1021/ct1006696
Assessing protein loop flexibility by hierarchical Monte Carlo sampling
Abstract
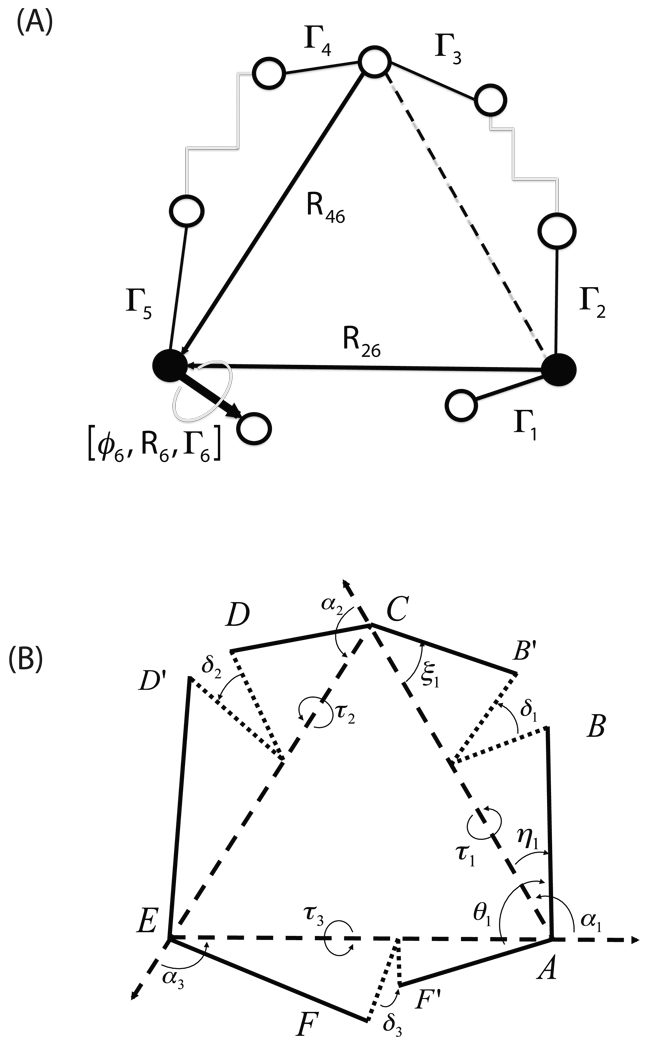
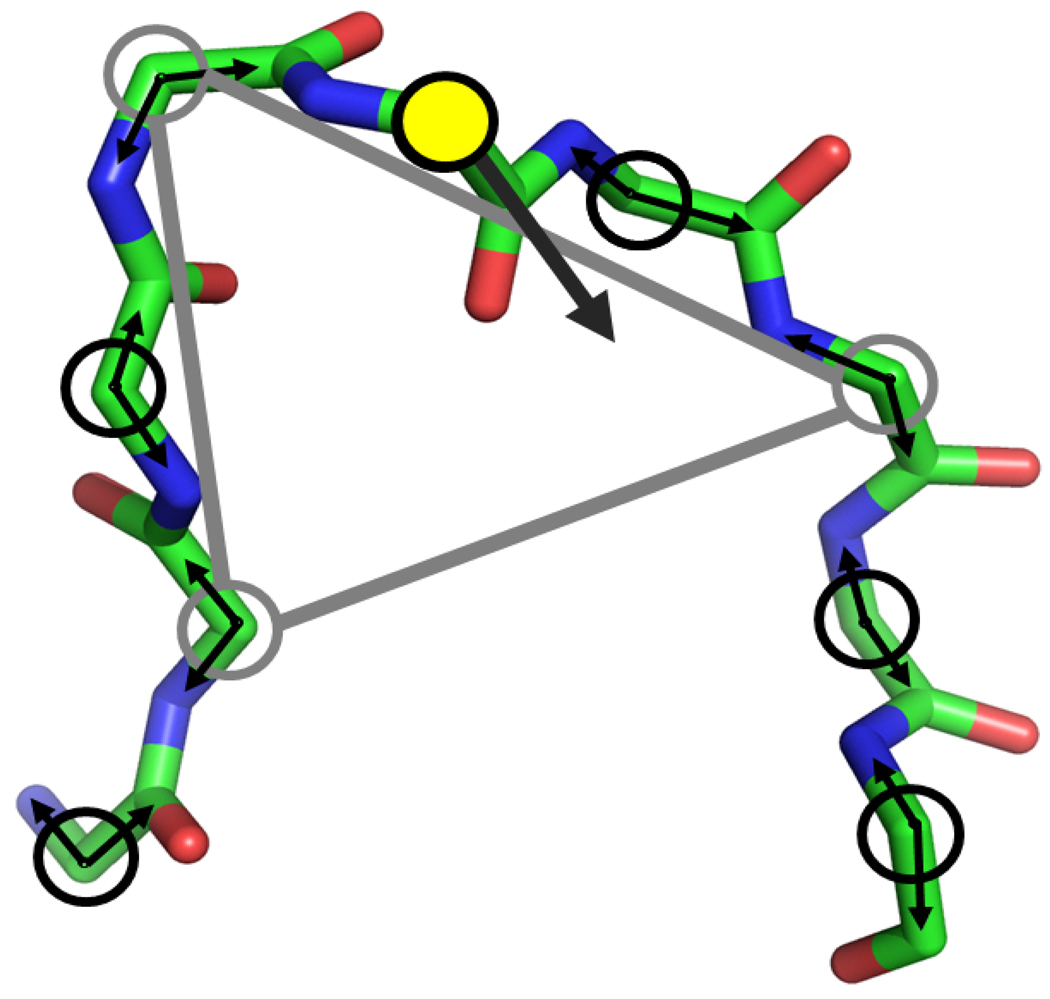
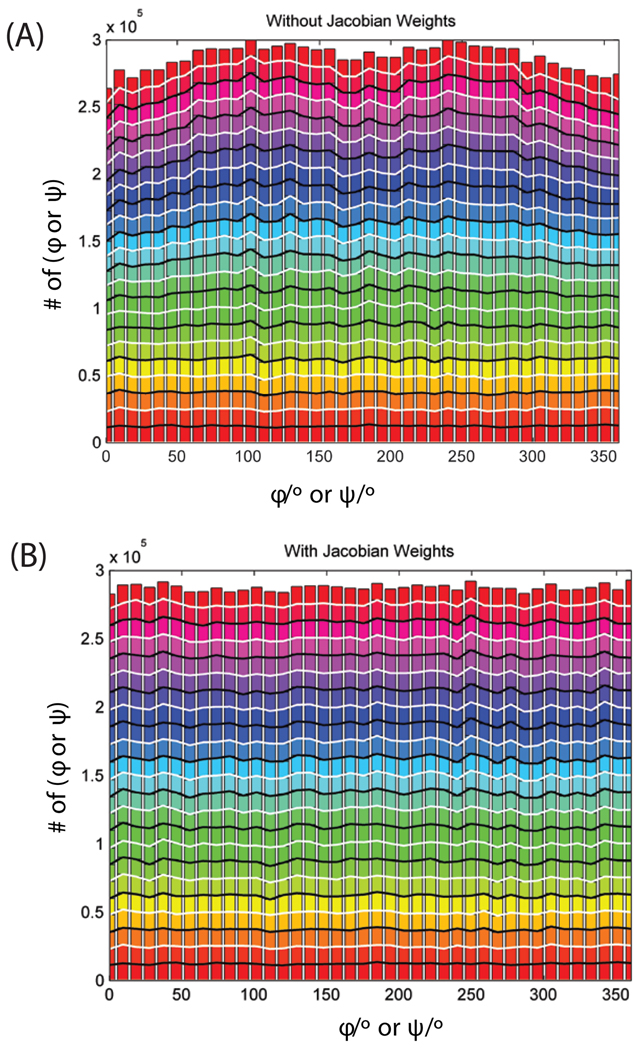
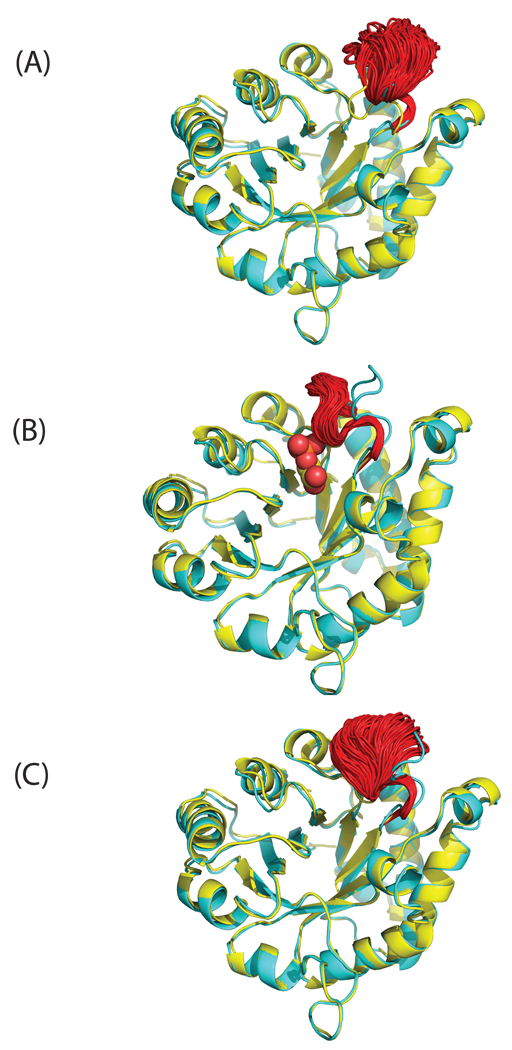
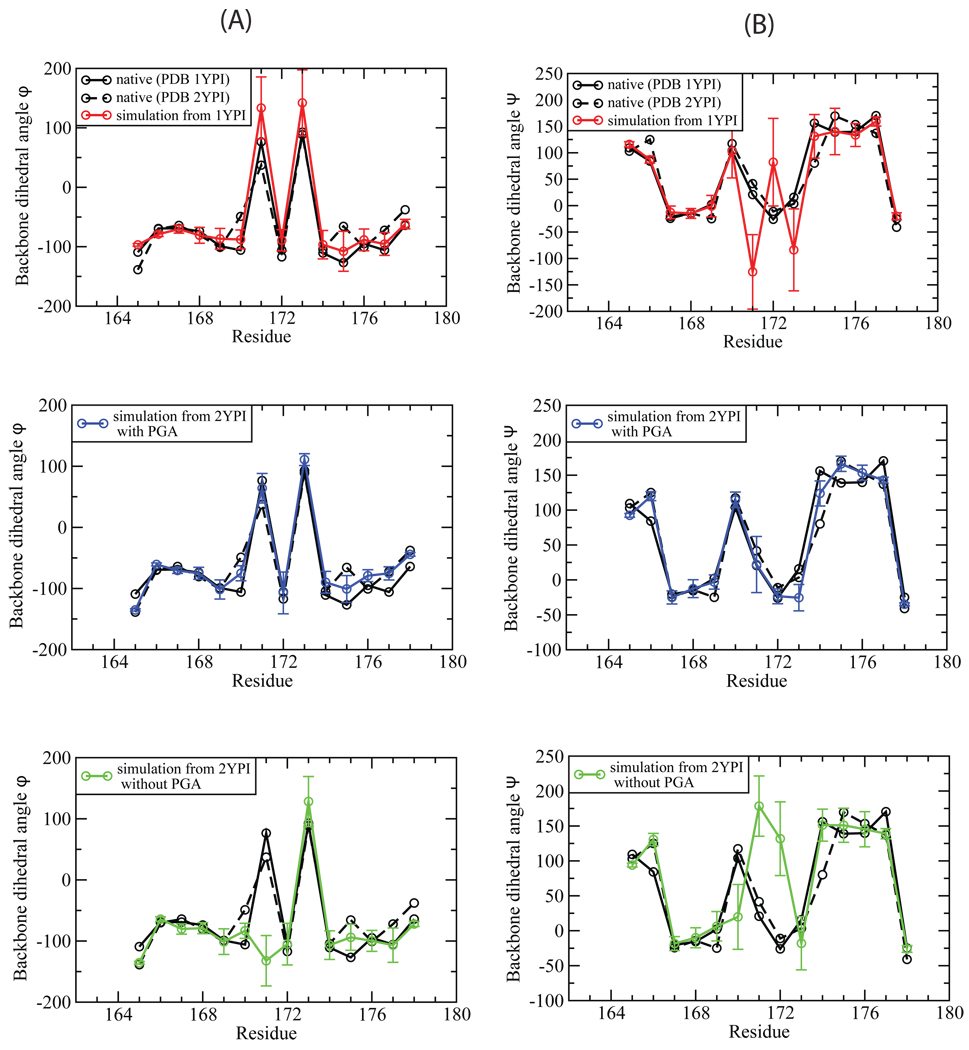
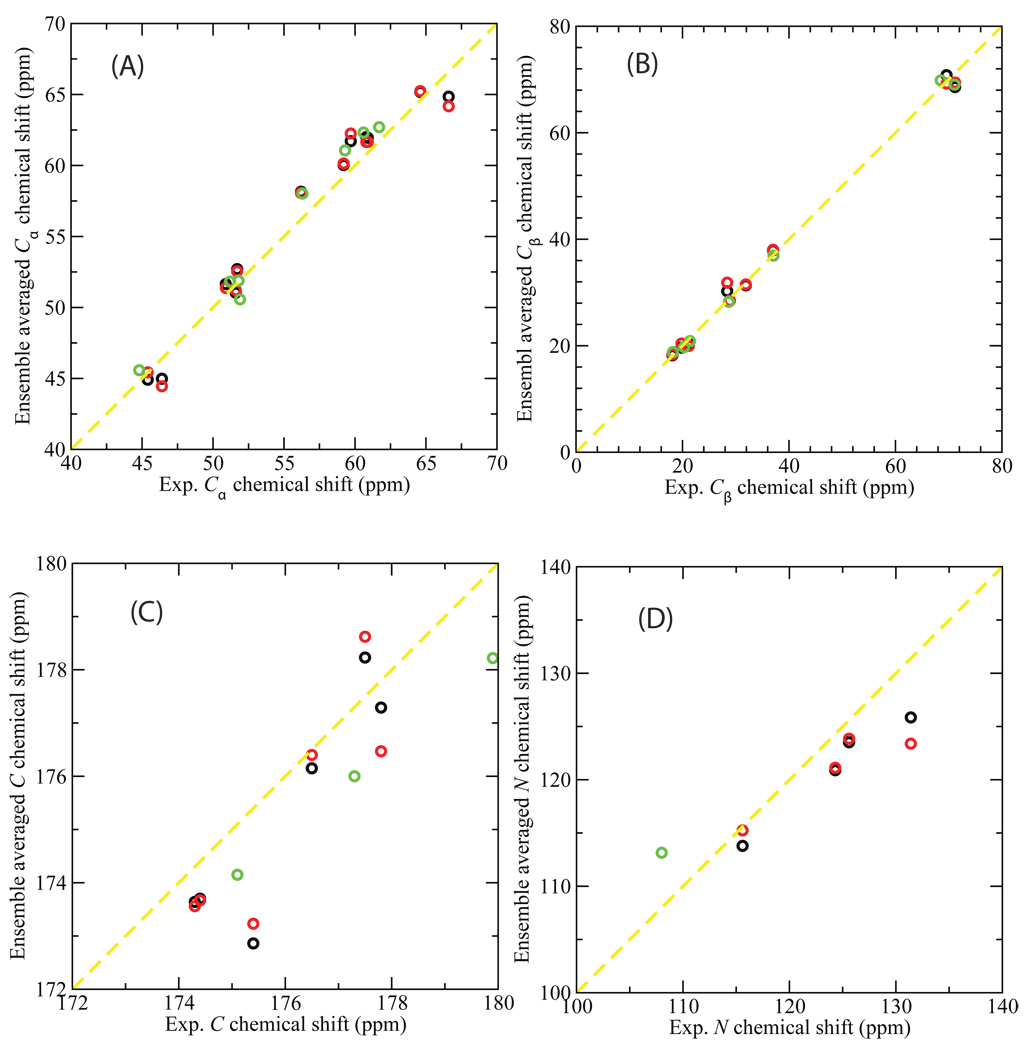
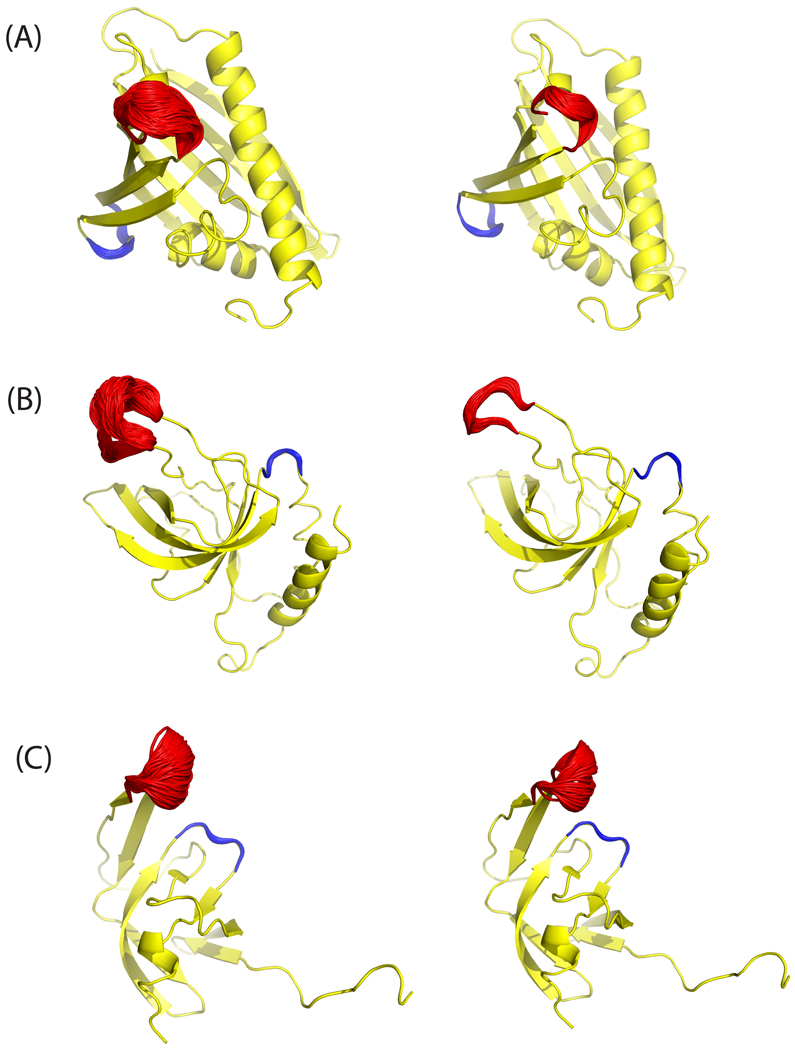
Loop flexibility is often crucial to protein biological function in solution. We report a new Monte Carlo method for generating conformational ensembles for protein loops and cyclic peptides. The approach incorporates the triaxial loop closure method which addresses the inverse kinematic problem for generating backbone move sets that do not break the loop. Sidechains are sampled together with the backbone in a hierarchical way, making it possible to make large moves that cross energy barriers. As an initial application, we apply the method to the flexible loop in triosephosphate isomerase that caps the active site, and demonstrate that the resulting loop ensembles agree well with key observations from previous structural studies. We also demonstrate, with 3 other test cases, the ability to distinguish relatively flexible and rigid loops within the same protein.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous