

On the scalability and requirements of whole protein mass spectrometry
- PMID: 21744800
- PMCID: PMC3165072
- DOI: 10.1021/ac2010795
On the scalability and requirements of whole protein mass spectrometry
Abstract
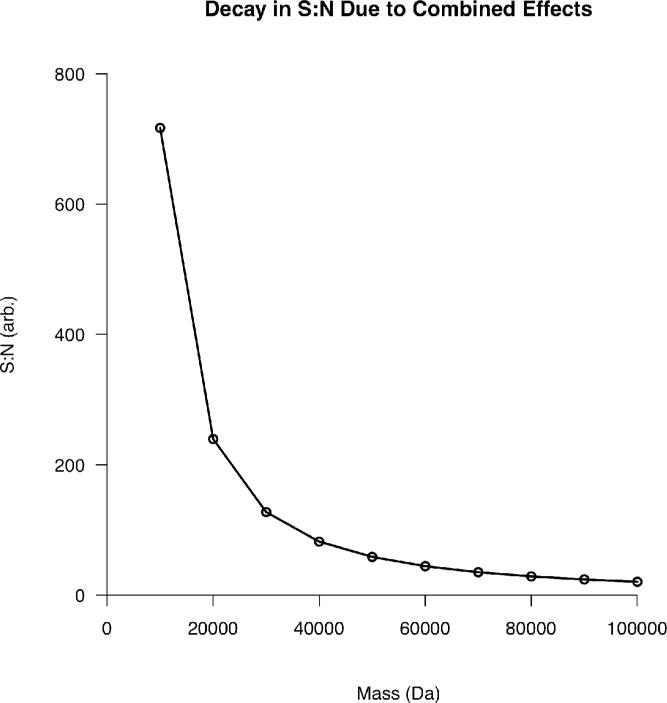
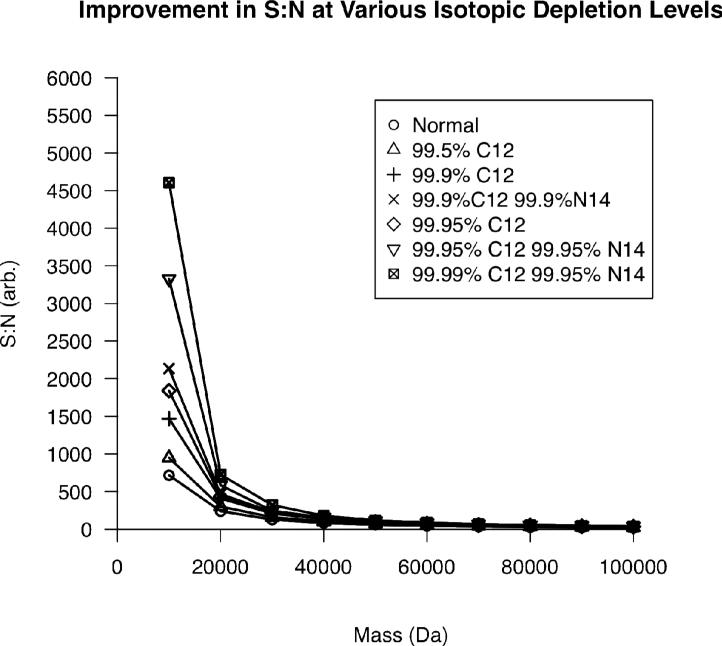
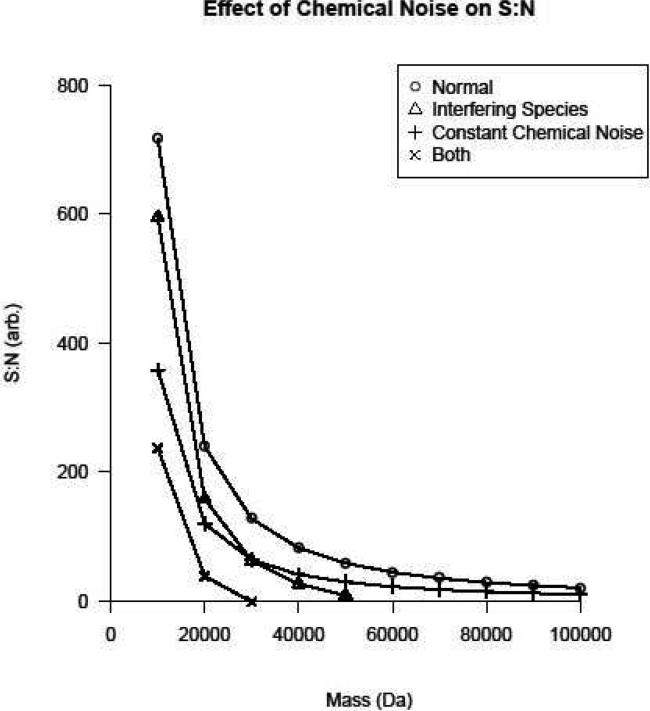
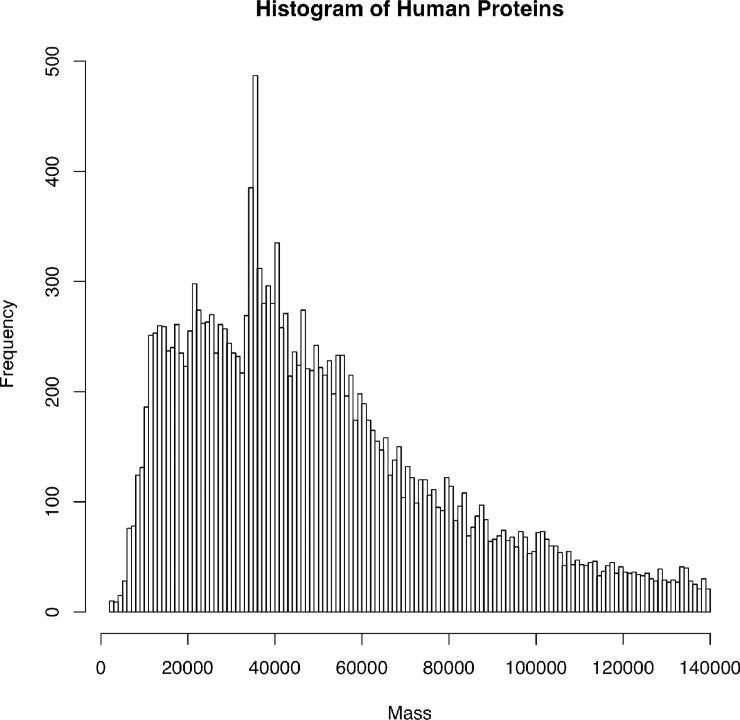
Top-down proteomics has improved over the past decade despite the significant challenges presented by the analysis of large protein ions. Here, the detection of these high mass species by electrospray-based mass spectrometry (MS) is examined from a theoretical perspective to understand the mass-dependent increases in the number of charge states, isotopic peaks, and interfering species present in typical protein mass spectra. Integrating these effects into a quantitative model captures the reduced ability to detect species over 25 kDa with the speed and sensitivity characteristic of proteomics based on <3 kDa peptide ions. The model quantifies the challenge that top-down proteomics faces with respect to current MS instrumentation and projects that depletion of (13)C and (15)N isotopes can improve detection at high mass by only <2-fold at 100 kDa whereas the effect is up to 5-fold at 10 kDa. Further, we find that supercharging electrosprayed proteins to the point of producing <5 charge states at high mass would improve detection by more than 20-fold.
Figures
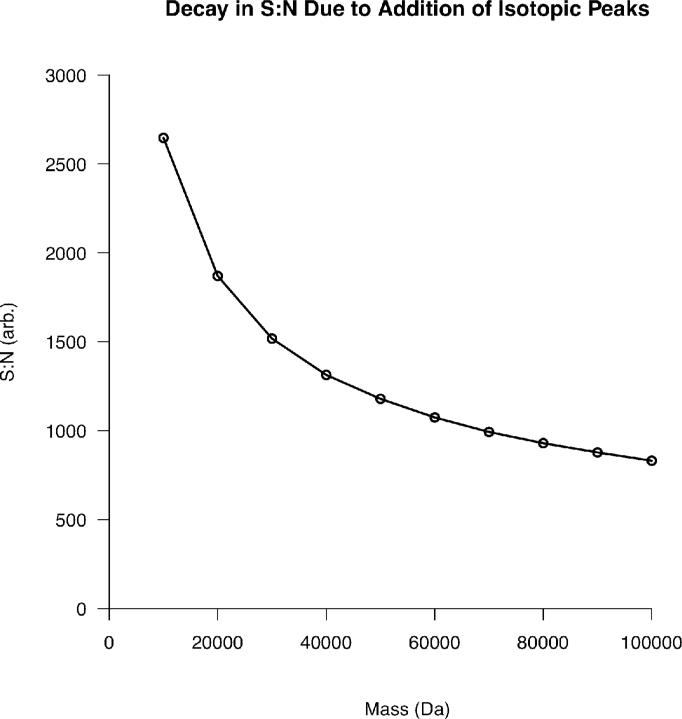
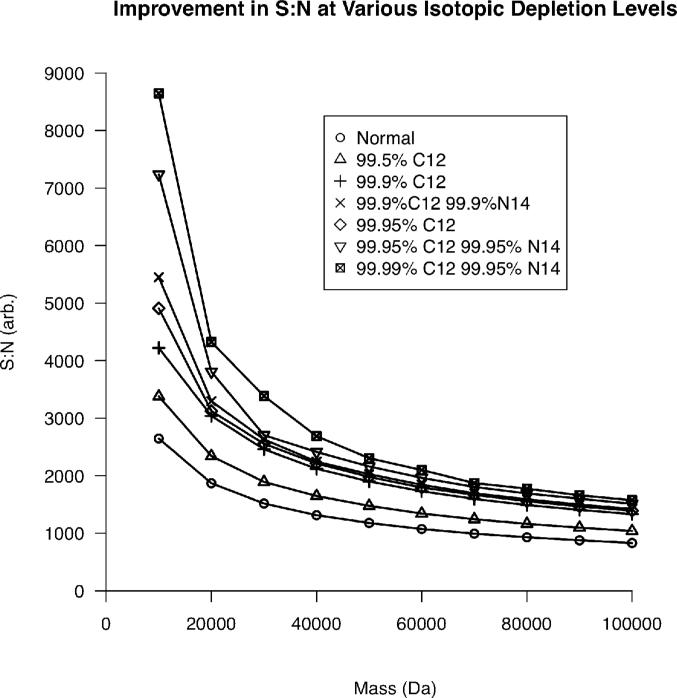
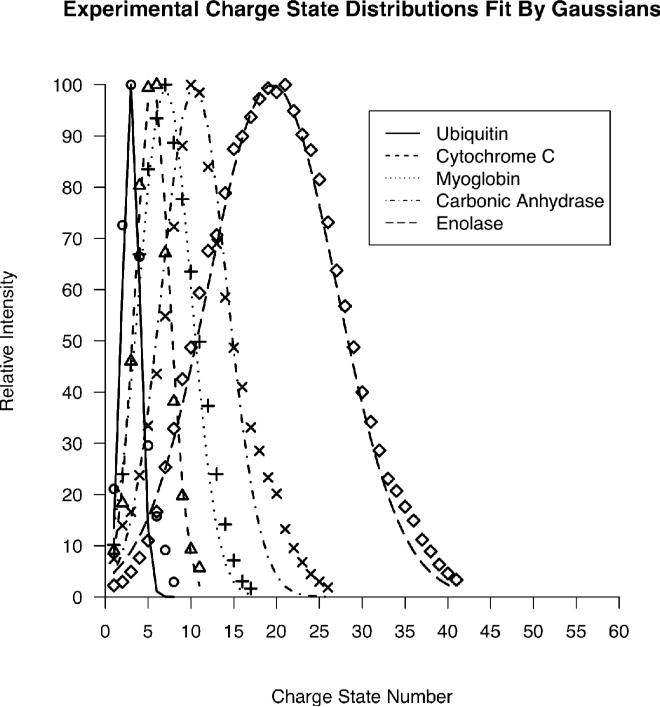
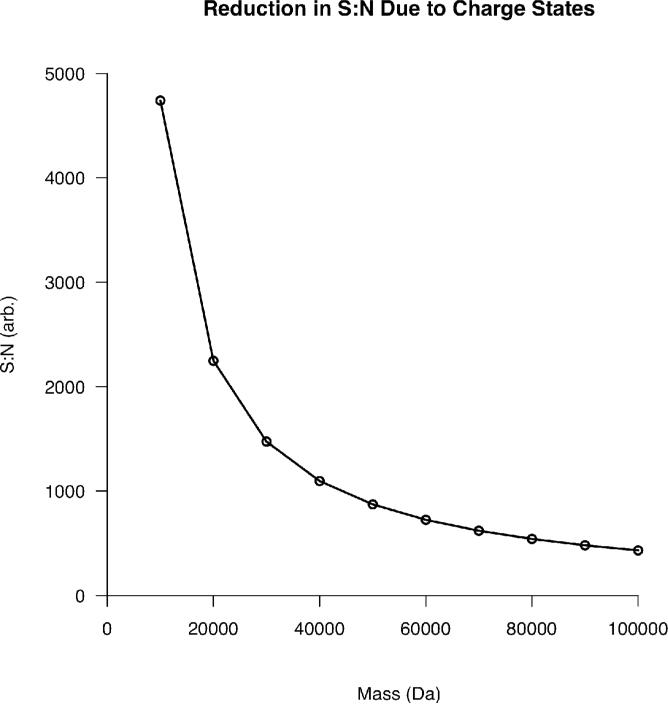
References
-
- Bunger MK, Cargile BJ, Ngunjiri A, Bundy JL, Stephenson JL., Jr. Anal Chem. 2008;80:1459–1467. - PubMed
-
- Meng F, Du Y, Miller LM, Patrie SM, Robinson DE, Kelleher NL. Anal Chem. 2004;76:2852–2858. - PubMed
-
- Sharma S, Simpson DC, Tolic N, Jaitly N, Mayampurath AM, Smith RD, Pasa-Tolic L. J Proteome Res. 2007;6:602–610. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
