

Solution structures of DEAD-box RNA chaperones reveal conformational changes and nucleic acid tethering by a basic tail
- PMID: 21746911
- PMCID: PMC3145681
- DOI: 10.1073/pnas.1109566108
Solution structures of DEAD-box RNA chaperones reveal conformational changes and nucleic acid tethering by a basic tail
Abstract
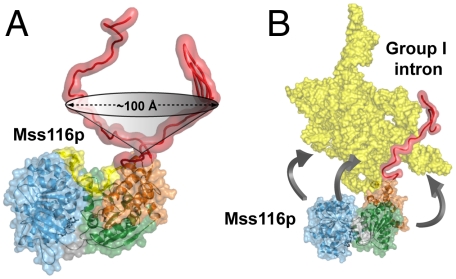
The mitochondrial DEAD-box proteins Mss116p of Saccharomyces cerevisiae and CYT-19 of Neurospora crassa are ATP-dependent helicases that function as general RNA chaperones. The helicase core of each protein precedes a C-terminal extension and a basic tail, whose structural role is unclear. Here we used small-angle X-ray scattering to obtain solution structures of the full-length proteins and a series of deletion mutants. We find that the two core domains have a preferred relative orientation in the open state without substrates, and we visualize the transition to a compact closed state upon binding RNA and adenosine nucleotide. An analysis of complexes with large chimeric oligonucleotides shows that the basic tails of both proteins are attached flexibly, enabling them to bind rigid duplex DNA segments extending from the core in different directions. Our results indicate that the basic tails of DEAD-box proteins contribute to RNA-chaperone activity by binding nonspecifically to large RNA substrates and flexibly tethering the core for the unwinding of neighboring duplexes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
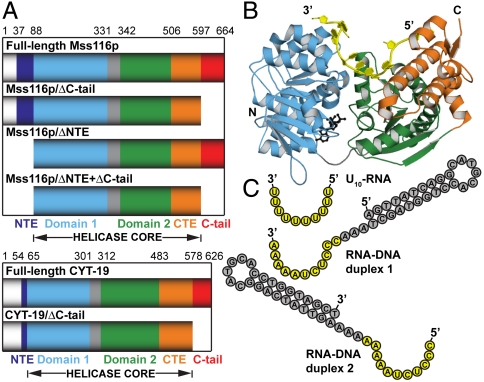
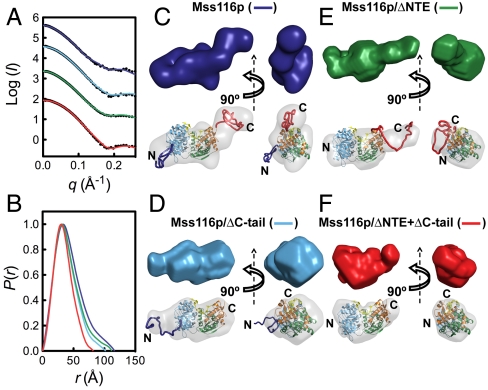
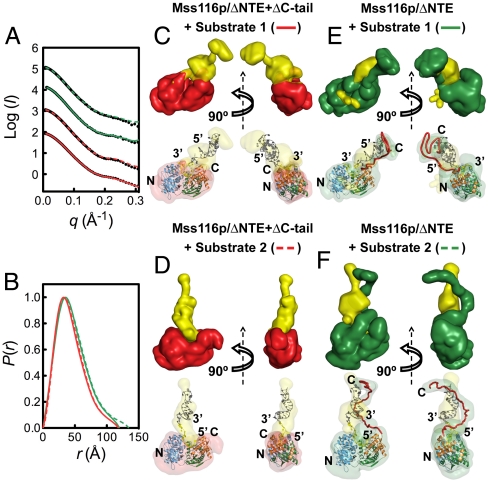
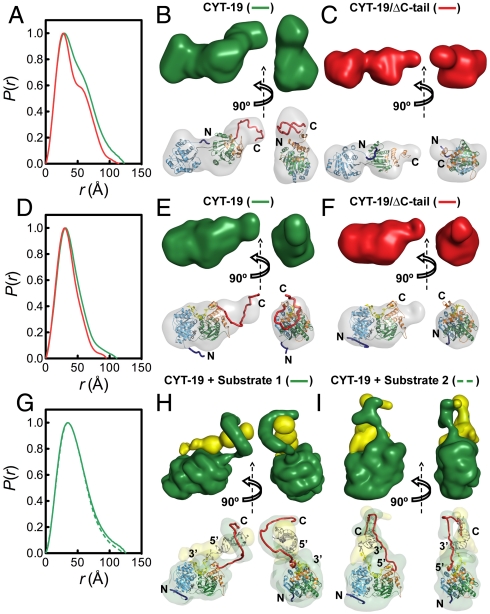
 are shown in yellow and black, respectively. (C) Schematic representations of nucleic acid substrates. RNA and DNA nucleotides are shown in yellow and gray, respectively, and nucleic-acid secondary structure was predicted using RNAfold (
are shown in yellow and black, respectively. (C) Schematic representations of nucleic acid substrates. RNA and DNA nucleotides are shown in yellow and gray, respectively, and nucleic-acid secondary structure was predicted using RNAfold (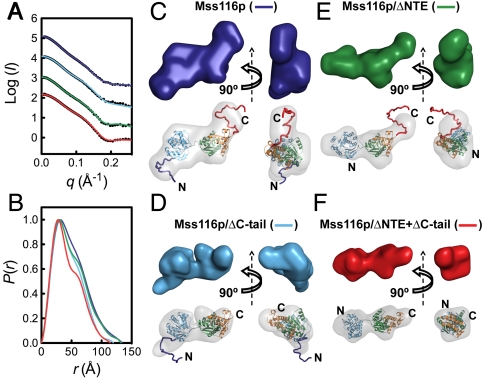
Similar articles
-
Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p.Nature. 2012 Oct 4;490(7418):121-5. doi: 10.1038/nature11402. Epub 2012 Sep 2. Nature. 2012. PMID: 22940866 Free PMC article.
-
Function of the C-terminal domain of the DEAD-box protein Mss116p analyzed in vivo and in vitro.J Mol Biol. 2008 Feb 1;375(5):1344-64. doi: 10.1016/j.jmb.2007.11.041. Epub 2007 Nov 22. J Mol Biol. 2008. PMID: 18096186 Free PMC article.
-
Molecular insights into RNA and DNA helicase evolution from the determinants of specificity for a DEAD-box RNA helicase.Elife. 2014 Dec 12;3:e04630. doi: 10.7554/eLife.04630. Elife. 2014. PMID: 25497230 Free PMC article.
-
Fluorescence methods in the investigation of the DEAD-box helicase mechanism.Exp Suppl. 2014;105:161-92. doi: 10.1007/978-3-0348-0856-9_8. Exp Suppl. 2014. PMID: 25095995 Review.
-
DEAD-box proteins as RNA helicases and chaperones.Wiley Interdiscip Rev RNA. 2011 Jan-Feb;2(1):135-52. doi: 10.1002/wrna.50. Wiley Interdiscip Rev RNA. 2011. PMID: 21297876 Free PMC article. Review.
Cited by
-
Characterization of the kinetics of RNA annealing and strand displacement activities of the E. coli DEAD-box helicase CsdA.RNA Biol. 2013 Jan;10(1):149-56. doi: 10.4161/rna.23475. Epub 2013 Jan 1. RNA Biol. 2013. PMID: 23291905 Free PMC article.
-
Structural basis for RNA-duplex recognition and unwinding by the DEAD-box helicase Mss116p.Nature. 2012 Oct 4;490(7418):121-5. doi: 10.1038/nature11402. Epub 2012 Sep 2. Nature. 2012. PMID: 22940866 Free PMC article.
-
Investigating increasingly complex macromolecular systems with small-angle X-ray scattering.IUCrJ. 2014 Oct 21;1(Pt 6):523-9. doi: 10.1107/S2052252514020843. eCollection 2014 Nov 1. IUCrJ. 2014. PMID: 25485132 Free PMC article. Review.
-
Iterative annealing mechanism explains the functions of the GroEL and RNA chaperones.Protein Sci. 2020 Feb;29(2):360-377. doi: 10.1002/pro.3795. Epub 2019 Dec 23. Protein Sci. 2020. PMID: 31800116 Free PMC article. Review.
-
Measuring the impact of cofactors on RNA helicase activities.Methods. 2022 Aug;204:376-385. doi: 10.1016/j.ymeth.2022.04.005. Epub 2022 Apr 14. Methods. 2022. PMID: 35429628 Free PMC article.
References
-
- Cordin O, Banroques J, Tanner NK, Linder P. The DEAD-box protein family of RNA helicases. Gene. 2006;367:17–37. - PubMed
-
- Jankowsky E, Fairman ME. RNA helicases—one fold for many functions. Curr Opin Struct Biol. 2007;17:316–324. - PubMed
-
- Hilbert M, Karow AR, Klostermeier D. The mechanism of ATP-dependent RNA unwinding by DEAD box proteins. Biol Chem. 2009;390:1237–1250. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
