

Primary hyperoxaluria
- PMID: 21748001
- PMCID: PMC3124893
- DOI: 10.4061/2011/864580
Primary hyperoxaluria
Abstract
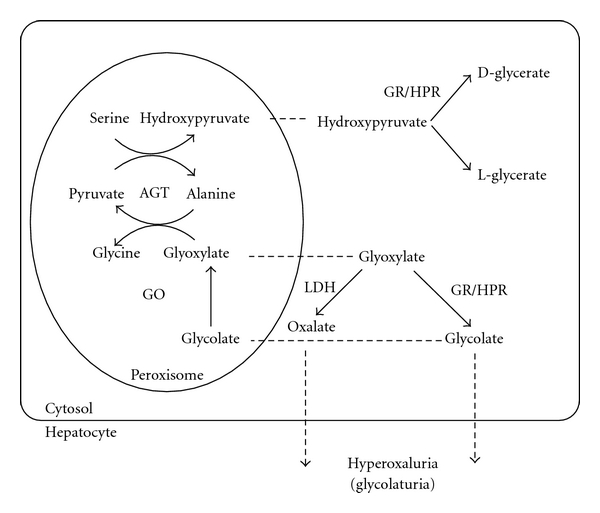
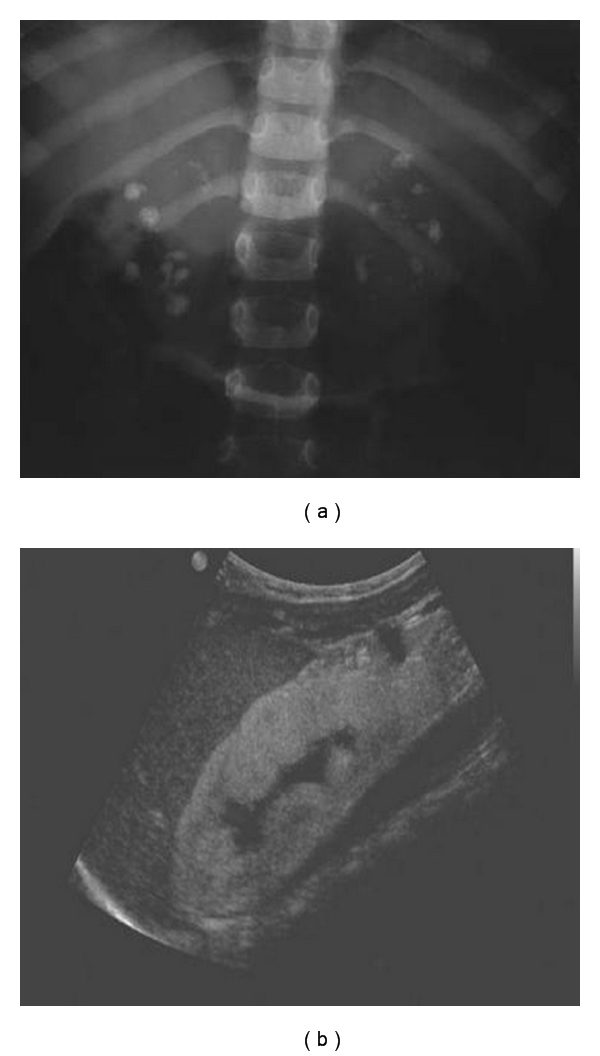
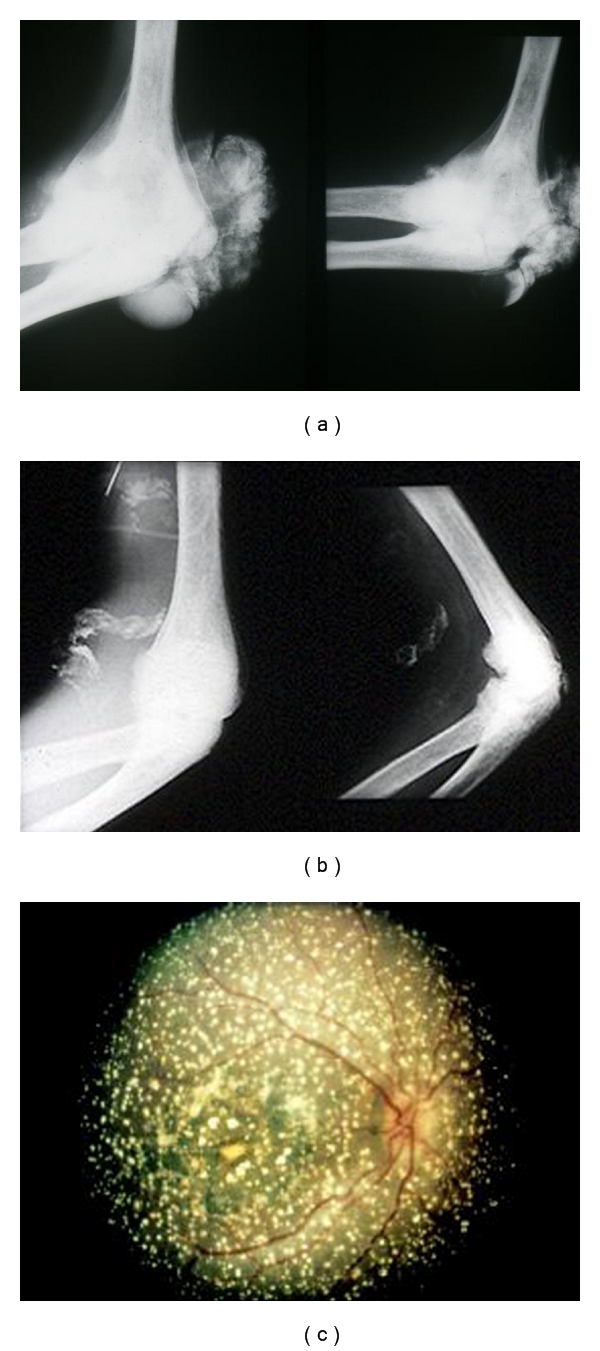
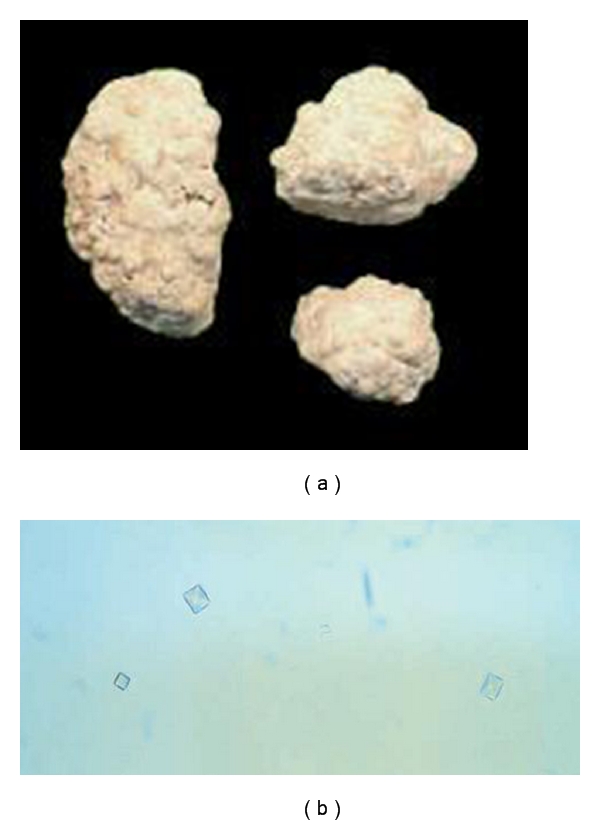
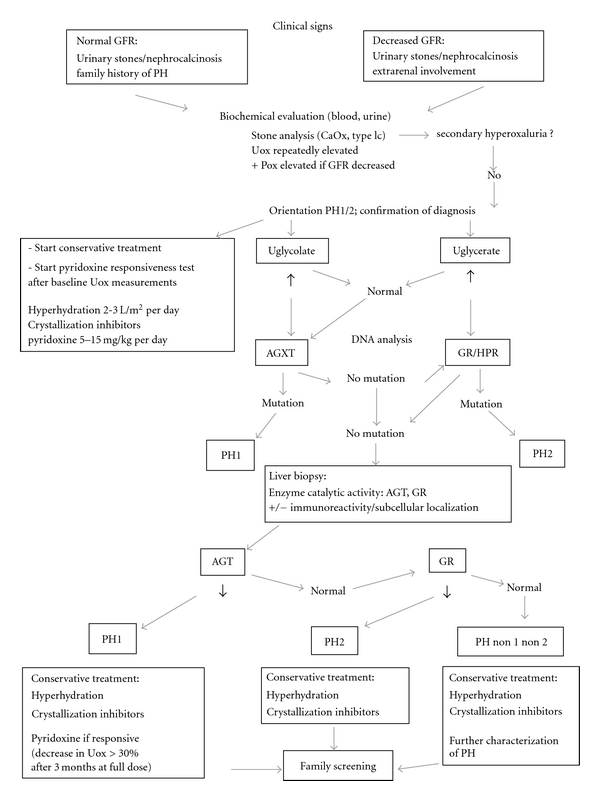
Primary hyperoxalurias (PH) are inborn errors in the metabolism of glyoxylate and oxalate. PH type 1, the most common form, is an autosomal recessive disorder caused by a deficiency of the liver-specific enzyme alanine, glyoxylate aminotransferase (AGT) resulting in overproduction and excessive urinary excretion of oxalate. Recurrent urolithiasis and nephrocalcinosis are the hallmarks of the disease. As glomerular filtration rate decreases due to progressive renal damage, oxalate accumulates leading to systemic oxalosis. Diagnosis is often delayed and is based on clinical and sonographic findings, urinary oxalate assessment, DNA analysis, and, if necessary, direct AGT activity measurement in liver biopsy tissue. Early initiation of conservative treatment, including high fluid intake, inhibitors of calcium oxalate crystallization, and pyridoxine in responsive cases, can help to maintain renal function in compliant subjects. In end-stage renal disease patients, the best outcomes have been achieved with combined liver-kidney transplantation which corrects the enzyme defect.
Figures
References
-
- Danpure CJ, Jennings PR. Peroxisomal alanine: glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Letters. 1986;201(1):20–24. - PubMed
-
- Purdue PE, Lumb MJ, Fox M, et al. Characterization and chromosomal mapping of a genomic clone encoding human alanine: glyoxylate aminotransferase. Genomics. 1991;10(1):34–42. - PubMed
-
- Mistry J, Danpure CJ, Chalmers RA. Hepatic D-glycerate dehydrogenase and glyoxylate reductase deficiency in primary hyperoxaluria type 2. Biochemical Society Transactions. 1988;16(4):626–627.
-
- Rumsby G, Cregeen DP. Identification and expression of a cDNA for human hydroxypyruvate/glyoxylate reductase. Biochimica et Biophysica Acta. 1999;1446(3):383–388. - PubMed
-
- Cramer SD, Ferree PM, Lin K, Milliner DS, Holmes RP. The gene encoding hydroxypyruvate reductase (GRHPR) is mutated in patients with primary hyperoxaluria type II. Human Molecular Genetics. 1999;8(11):2063–2069. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
