

Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project
- PMID: 21749982
- PMCID: PMC3356493
- DOI: 10.1136/gut.2010.236026
Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project
Abstract
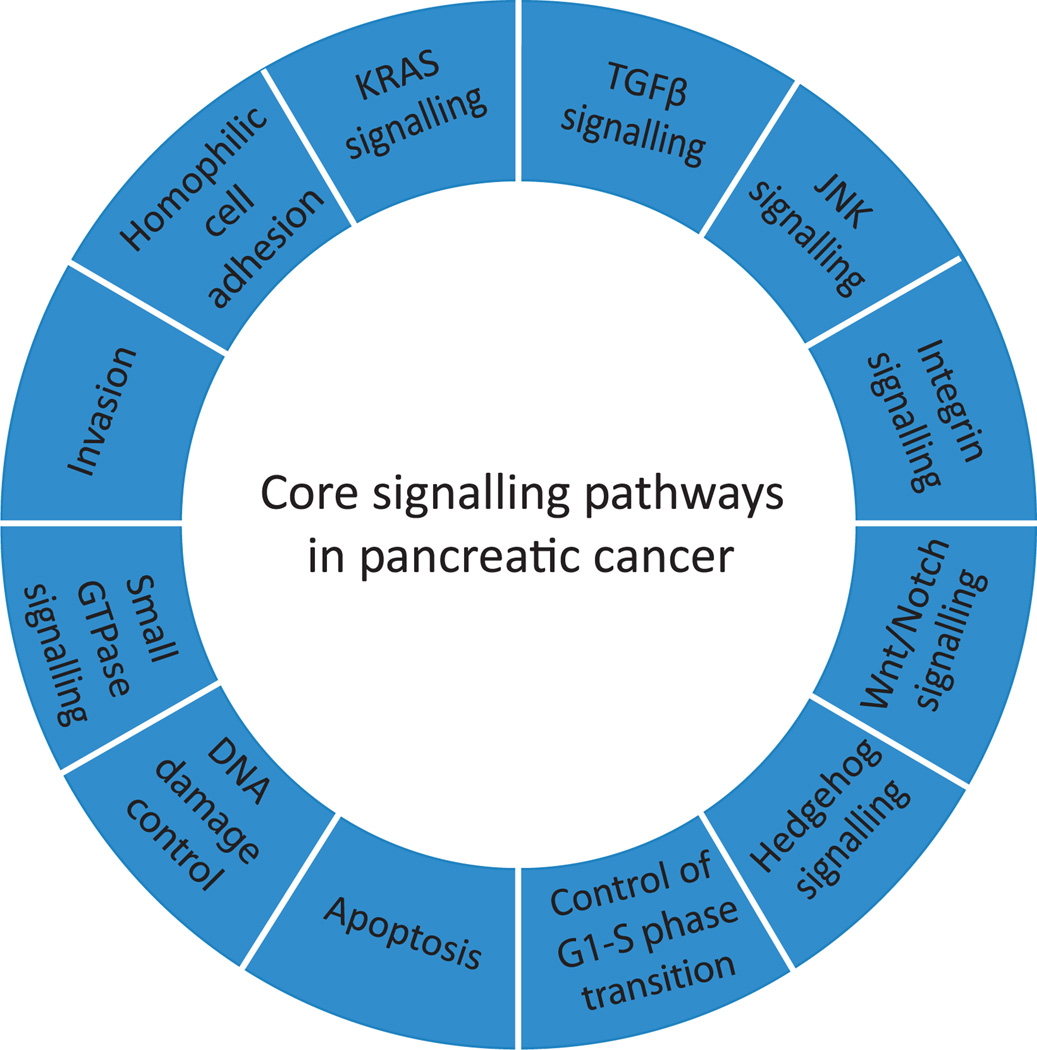
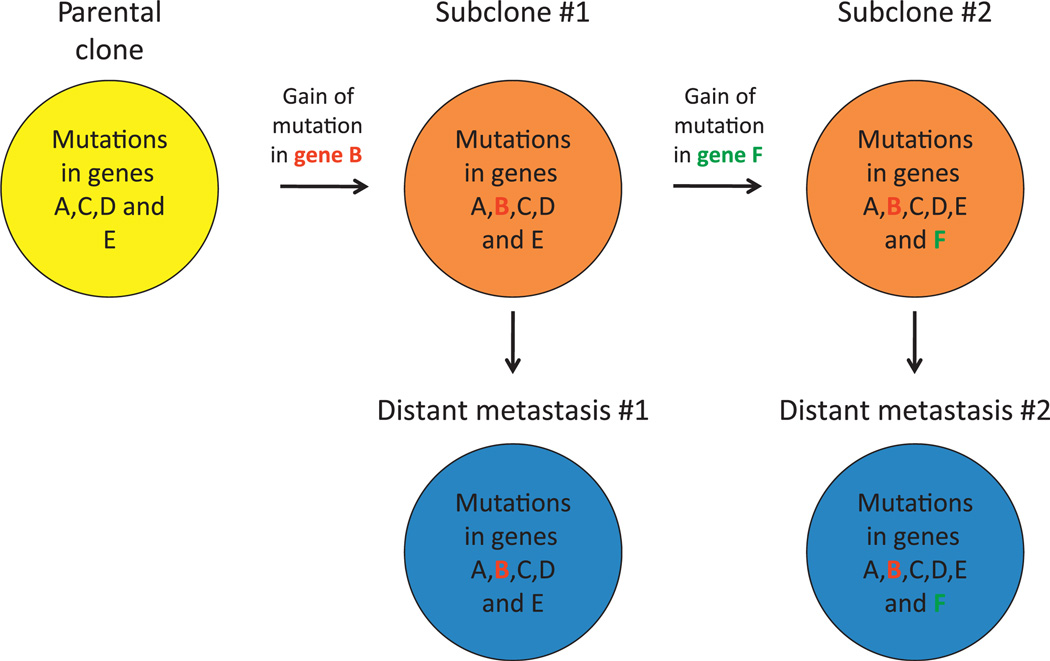
Pancreatic cancer is a disease caused by the accumulation of genetic alterations in specific genes. Elucidation of the human genome sequence, in conjunction with technical advances in the ability to perform whole exome sequencing, have provided new insight into the mutational spectra characteristic of this lethal tumour type. Most recently, exomic sequencing has been used to clarify the clonal evolution of pancreatic cancer as well as provide time estimates of pancreatic carcinogenesis, indicating that a long window of opportunity may exist for early detection of this disease while in the curative stage. Moving forward, these mutational analyses indicate potential targets for personalised diagnostic and therapeutic intervention as well as the optimal timing for intervention based on the natural history of pancreatic carcinogenesis and progression.
Conflict of interest statement
There are no relevant financial conflicts to disclose.
Figures



References
-
- Jemal A, Siegel R, Xu J, et al. Cancer statistics, 2010. CA Cancer J clin. 2010;60:277–300. - PubMed
-
- Malvezzi M, Arfe A, Bertuccio P, et al. European cancer mortality predictions for the year 2011. Ann Oncol. 2011;22:947–956. - PubMed
-
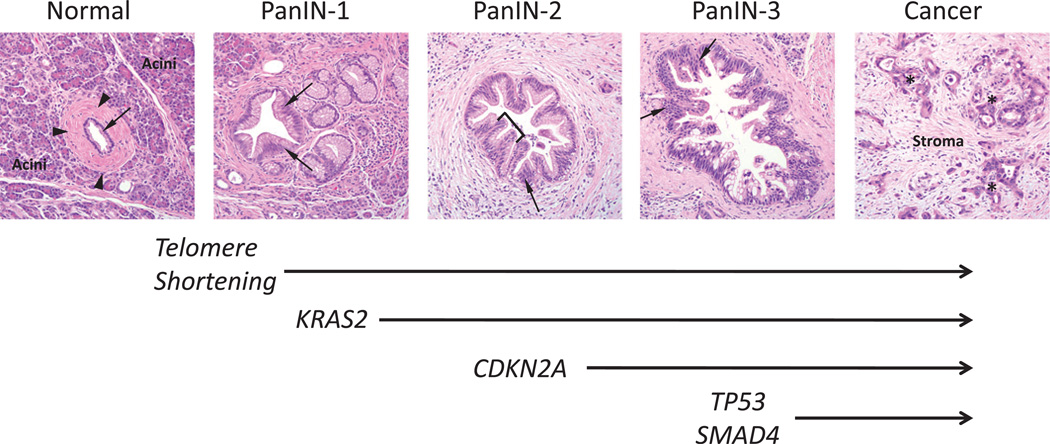
- Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997;57:2140–2143. - PubMed
-
- Wilentz RE, Iacobuzio-Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000;60:2002–2006. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical