

Loss of exon identity is a common mechanism of human inherited disease
- PMID: 21750108
- PMCID: PMC3202274
- DOI: 10.1101/gr.118638.110
Loss of exon identity is a common mechanism of human inherited disease
Abstract
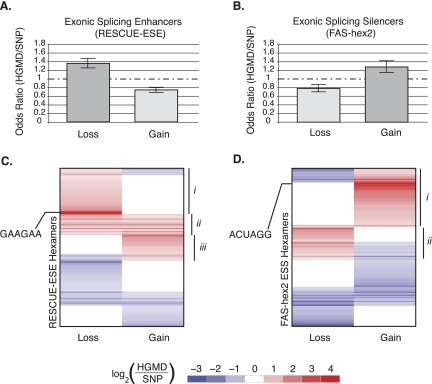
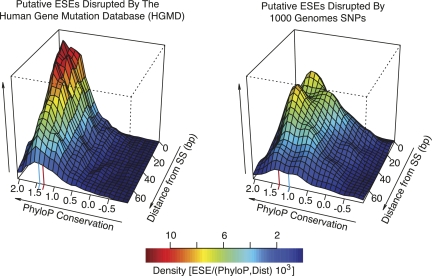
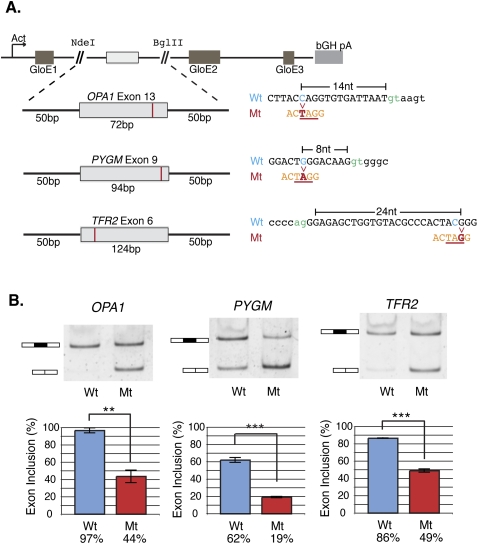
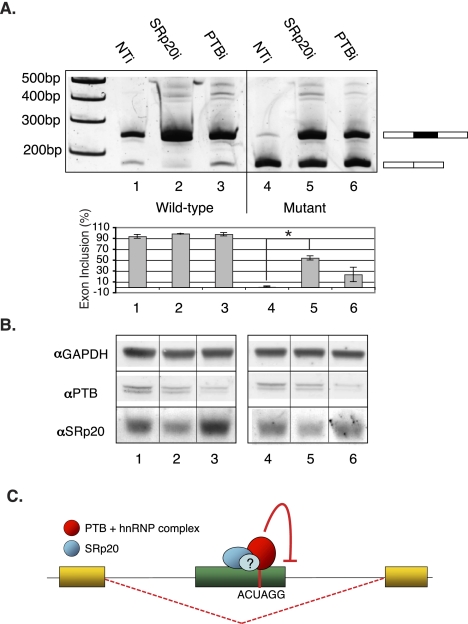
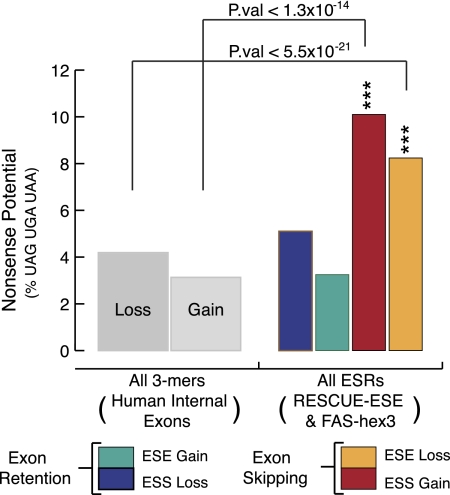
It is widely accepted that at least 10% of all mutations causing human inherited disease disrupt splice-site consensus sequences. In contrast to splice-site mutations, the role of auxiliary cis-acting elements such as exonic splicing enhancers (ESE) and exonic splicing silencers (ESS) in human inherited disease is still poorly understood. Here we use a top-down approach to determine rates of loss or gain of known human exonic splicing regulatory (ESR) sequences associated with either disease-causing mutations or putatively neutral single nucleotide polymorphisms (SNPs). We observe significant enrichment toward loss of ESEs and gain of ESSs among inherited disease-causing variants relative to neutral polymorphisms, indicating that exon skipping may play a prominent role in aberrant gene regulation. Both computational and biochemical approaches underscore the relevance of exonic splicing enhancer loss and silencer gain in inherited disease. Additionally, we provide direct evidence that both SRp20 (SRSF3) and possibly PTB (PTBP1) are involved in the function of a splicing silencer that is created de novo by a total of 83 different inherited disease mutations in 67 different disease genes. Taken together, we find that ~25% (7154/27,681) of known mis-sense and nonsense disease-causing mutations alter functional splicing signals within exons, suggesting a much more widespread role for aberrant mRNA processing in causing human inherited disease than has hitherto been appreciated.
Figures
References
-
- Barash Y, Calarco JA, Gao W, Pan Q, Wang X, Shai O, Blencowe BJ, Frey BJ 2010. Deciphering the splicing code. Nature 465: 53–59 - PubMed
-
- Benjamini Y, Hochberg Y 1995. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol 57: 289–300
-
- Biasiotto G, Camaschella C, Forni GL, Polotti A, Zecchina G, Arosio P 2008. New TFR2 mutations in young Italian patients with hemochromatosis. Haematologica 93: 309–310 - PubMed
-
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis KD, Ovcharenko I, Pachter L, Rubin EM 2003. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science 299: 1391–1394 - PubMed
-
- Bruno C, Tamburino L, Kawashima N, Andreu AL, Shanske S, Hadjigeorgiou GM, Kawashima A, DiMauro S 1999. A nonsense mutation in the myophosphorylase gene in a Japanese family with McArdle's disease. Neuromuscul Disord 9: 34–37 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous