

iPS cells to model CDKL5-related disorders
- PMID: 21750574
- PMCID: PMC3218106
- DOI: 10.1038/ejhg.2011.131
iPS cells to model CDKL5-related disorders
Abstract
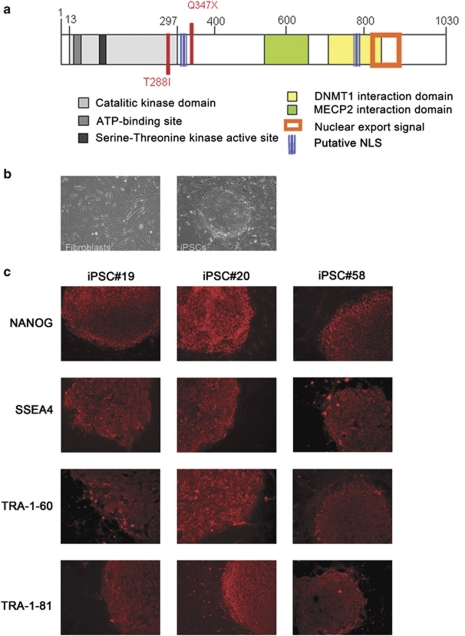
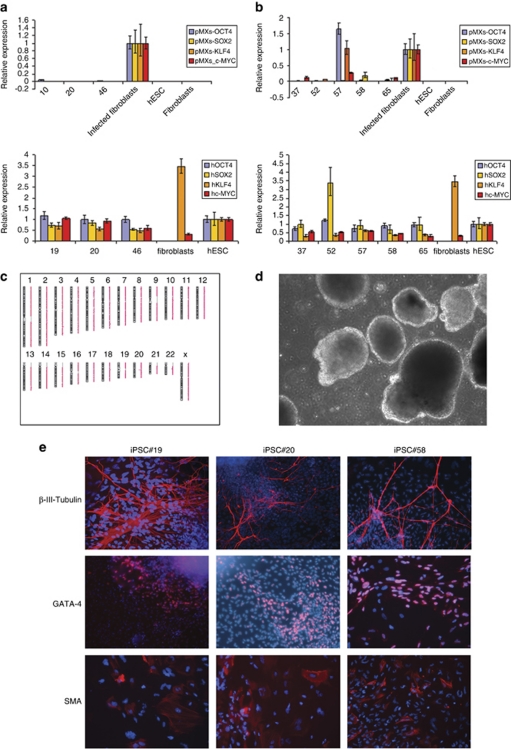
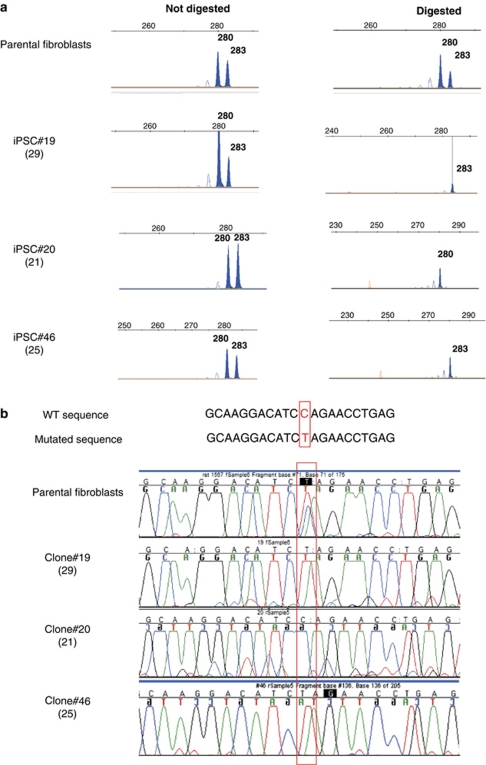
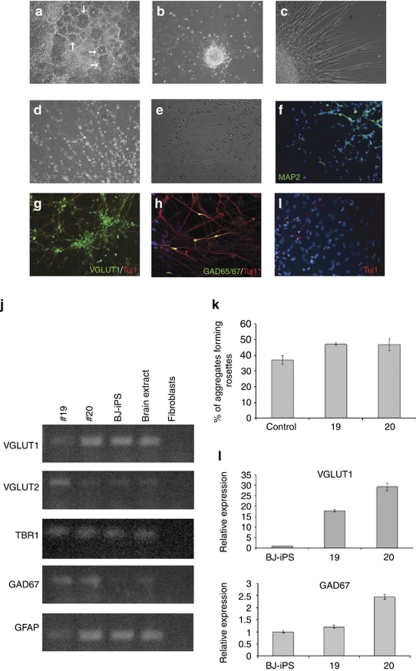
Rett syndrome (RTT) is a progressive neurologic disorder representing one of the most common causes of mental retardation in females. To date mutations in three genes have been associated with this condition. Classic RTT is caused by mutations in the MECP2 gene, whereas variants can be due to mutations in either MECP2 or FOXG1 or CDKL5. Mutations in CDKL5 have been identified both in females with the early onset seizure variant of RTT and in males with X-linked epileptic encephalopathy. CDKL5 is a kinase protein highly expressed in neurons, but its exact function inside the cell is unknown. To address this issue we established a human cellular model for CDKL5-related disease using the recently developed technology of induced pluripotent stem cells (iPSCs). iPSCs can be expanded indefinitely and differentiated in vitro into many different cell types, including neurons. These features make them the ideal tool to study disease mechanisms directly on the primarily affected neuronal cells. We derived iPSCs from fibroblasts of one female with p.Q347X and one male with p.T288I mutation, affected by early onset seizure variant and X-linked epileptic encephalopathy, respectively. We demonstrated that female CDKL5-mutated iPSCs maintain X-chromosome inactivation and clones express either the mutant CDKL5 allele or the wild-type allele that serve as an ideal experimental control. Array CGH indicates normal isogenic molecular karyotypes without detection of de novo CNVs in the CDKL5-mutated iPSCs. Furthermore, the iPS cells can be differentiated into neurons and are thus suitable to model disease pathogenesis in vitro.
Figures
References
-
- Chahrour M, Zoghbi HY. The story of Rett syndrome: from clinic to neurobiology. Neuron. 2007;56:422–437. - PubMed
-
- Hagberg BA, Skjeldal OH. Rett variants: a suggested model for inclusion criteria. Pediatr Neurol. 1994;11:5–11. - PubMed
-
- Rolando S. Rett syndrome: report of eight cases. Brain Dev. 1985;7:290–296. - PubMed
-
- Hanefeld F. The clinical pattern of the Rett syndrome. Brain Dev. 1985;7:320–325. - PubMed
-
- Zappella M. The preserved speech variant of the Rett complex: a report of 8 cases. Eur Child Adolesc Psychiatry. 1997;6:23–25. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
