

Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: an in vivo study
- PMID: 21750656
- PMCID: PMC3132848
- DOI: 10.1593/neo.11134
Metabolic targeting of lactate efflux by malignant glioma inhibits invasiveness and induces necrosis: an in vivo study
Abstract
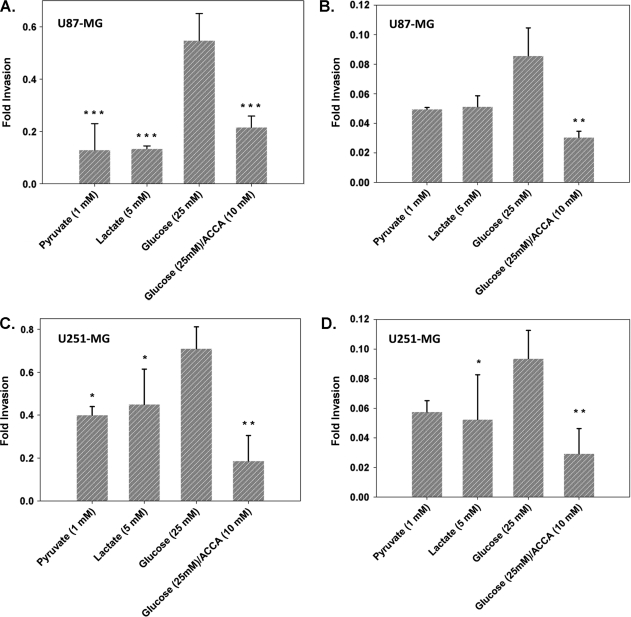
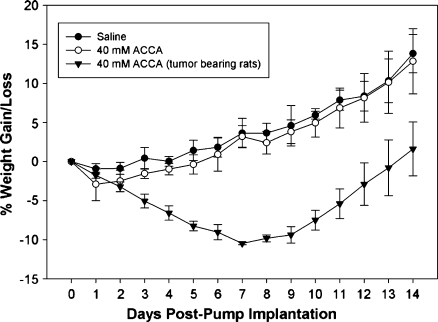
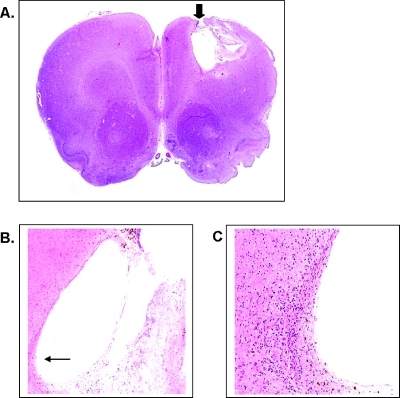
Glioblastoma multiforme (GBM) are the most malignant among brain tumors. They are frequently refractory to chemotherapy and radiotherapy with mean patient survival of approximately 6 months, despite surgical intervention. The highly glycolytic nature of glioblastomas describes their propensity to metabolize glucose to lactic acid at an elevated rate. To survive, GBMs efflux lactic acid to the tumor microenvironment through transmembrane transporters denoted monocarboxylate transporters (MCTs). We hypothesized that inhibition of MCT function would impair the glycolytic metabolism and affect both glioma invasiveness and survival. We examined the effect on invasiveness with α-cyano-4-hydroxy-cinnamic acid (ACCA, 4CIN, CHCA), a small-molecule inhibitor of lactate transport, through Matrigel-based and organotypic (brain) slice culture invasive assays using U87-MG and U251-MG glioma cells. We then conducted studies in immunodeficient rats by stereotaxic intracranial implantation of the glioma cells followed by programmed orthotopic application of ACCA through osmotic pumps. Effect on the implanted tumor was monitored by small-animal magnetic resonance imaging. Our assays indicated that glioma invasion was markedly impaired when lactate efflux was inhibited. Convection-enhanced delivery of inhibitor to the tumor bed caused tumor necrosis, with 50% of the animals surviving beyond the experimental end points (3 months after inhibitor exhaustion). Most importantly, control animals did not display any adverse neurologic effects during orthotopic administration of ACCA to brain through programmed delivery. These results indicate the clinical potential of targeting lactate efflux in glioma through delivery of small-molecule inhibitors of MCTs either to the tumor bed or to the postsurgical resection cavity.
Figures





References
-
- Warburg O, Dickens F Kaiser Wilhelm-Institut fur Biologie B, author. The Metabolism of Tumours: Investigations from the Kaiser-Wilhelm Institute for Biology, Berlin-Dahlem. London, UK: Constable & Co, Ltd; 1930.
-
- Baggetto LG. Deviant energetic metabolism of glycolytic cancer cells. Biochimie. 1992;74:959–974. - PubMed
-
- Pedersen PL. Tumor mitochondria and the bioenergetics of cancer cells. Prog Exp Tumor Res. 1978;22:190–274. - PubMed
-
- Walenta S, Schroeder T, Mueller-Klieser W. Lactate in solid malignant tumors: potential basis of a metabolic classification in clinical oncology. Curr Med Chem. 2004;11:2195–2204. - PubMed
-
- Walenta S, Mueller-Klieser WF. Lactate: mirror and motor of tumor malignancy. Semin Radiat Oncol. 2004;14:267–274. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous