

Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism
- PMID: 21753784
- PMCID: PMC3193573
- DOI: 10.1038/jid.2011.189
Pathogenesis-based therapy reverses cutaneous abnormalities in an inherited disorder of distal cholesterol metabolism
Abstract
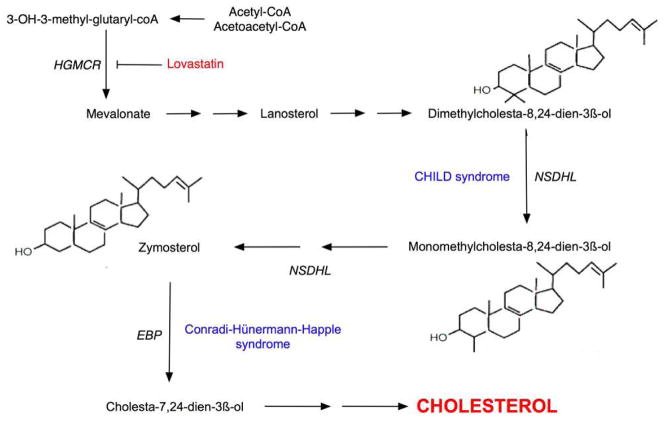
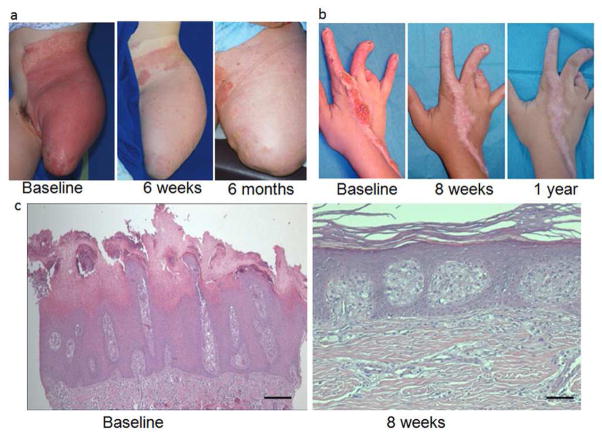
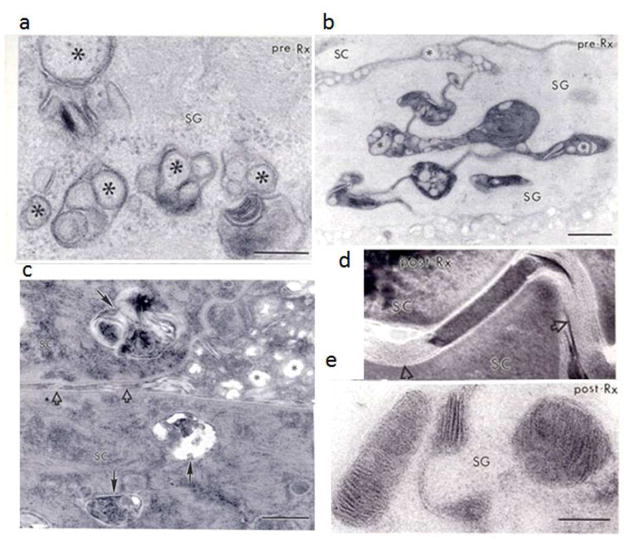
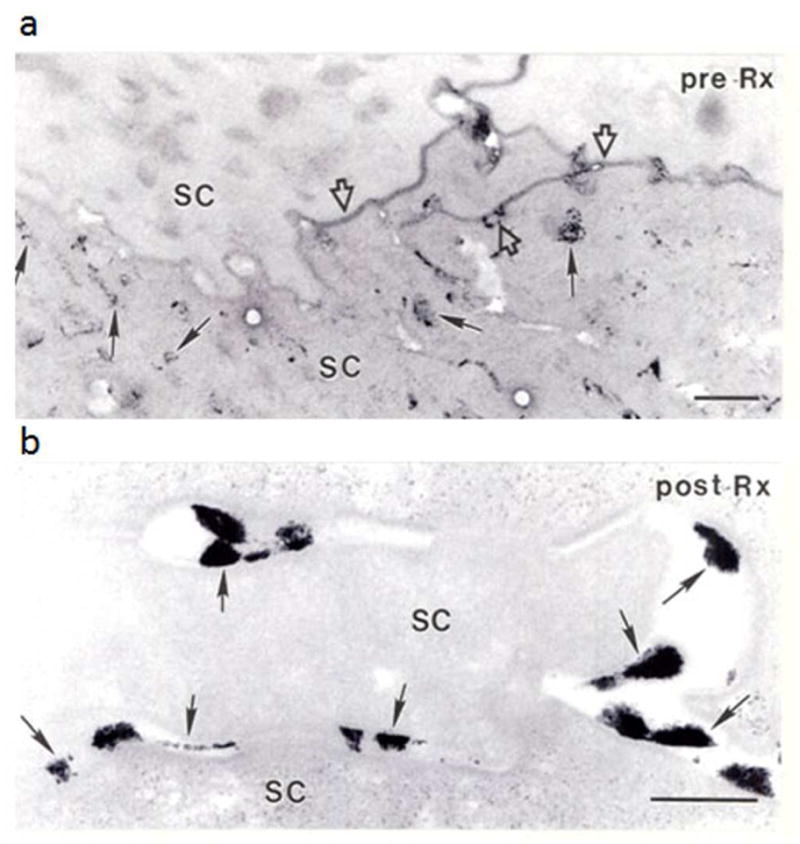
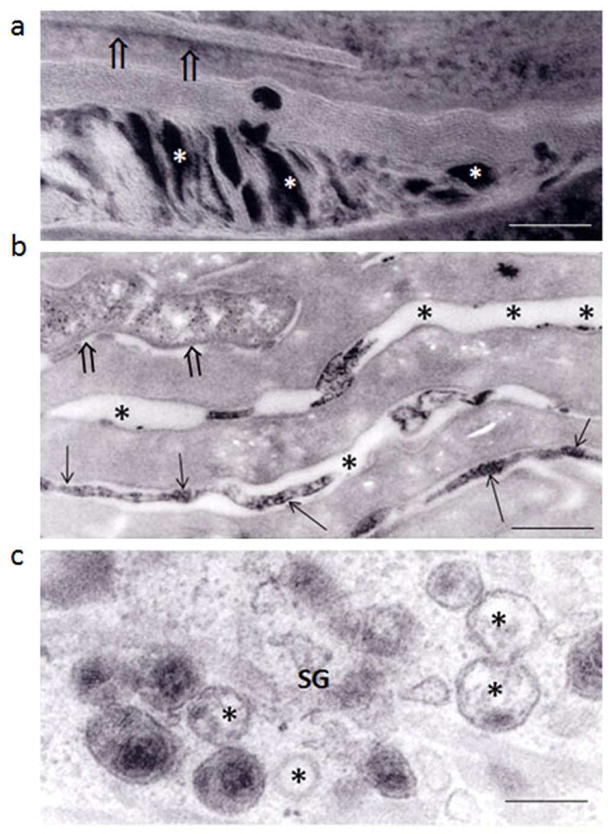
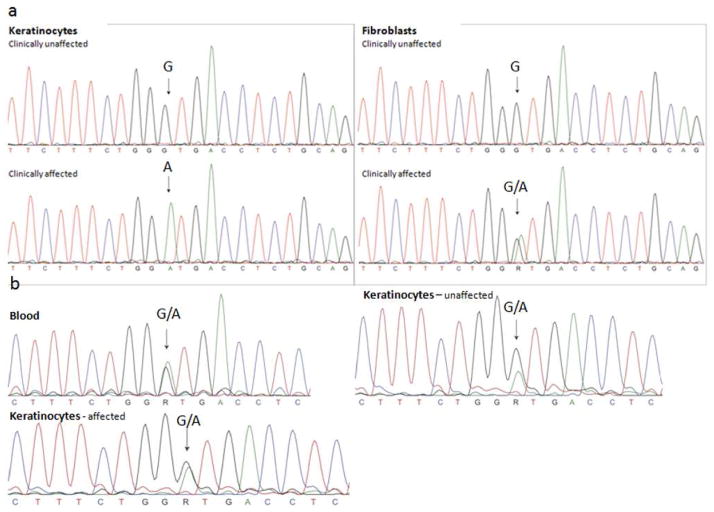
Identification of the underlying genetic, cellular, and biochemical basis of lipid metabolic disorders provides an opportunity to deploy corrective, mechanism-targeted, topical therapy. We assessed this therapeutic approach in two patients with Congenital Hemidysplasia with Ichthyosiform erythroderma and Limb Defects (CHILD) syndrome, an X-linked dominant disorder of distal cholesterol metabolism. On the basis of the putative pathogenic role of both pathway-product deficiency of cholesterol and accumulation of toxic metabolic intermediates, we assessed the efficacy of combined therapy with lovastatin and cholesterol. We also evaluated the basis for the poorly understood, unique lateralization of the cutaneous and bone malformations of CHILD syndrome by analyzing gene activation in abnormal and unaffected skin. Ultrastructural analysis of affected skin showed evidence of both cholesterol depletion and toxic metabolic accumulation. Topical treatment with lovastatin/cholesterol (but not cholesterol alone) virtually cleared skin lesions by 3 months, accompanied by histological and ultrastructural normalization of epidermal structure and lipid secretion. The unusual lateralization of abnormalities in CHILD syndrome reflects selective clearance of keratinocytes and fibroblasts that express the mutant allele from the unaffected side. These findings validate pathogenesis-based therapy that provides the deficient end product and prevents accumulation of toxic metabolites, an approach of potential utility for other syndromic lipid metabolic disorders.
Conflict of interest statement
Conflict of Interest
The authors state no conflict of interest.
Figures
References
-
- Akiyama M, Sakai K, Hayasaka K, et al. Conradi-Hunermann-Happle syndrome with abnormal lamellar granule contents. Br J Dermatol. 2009;160:1335–7. - PubMed
-
- Chiang C, Swan RZ, Grachtchouk M, et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev Biol. 1999;205:1–9. - PubMed
-
- Cooper MK, Wassif CA, Krakowiak PA, et al. A defective response to Hedgehog signaling in disorders of cholesterol biosynthesis. Nat Genet. 2003;33:508–13. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
