

Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing
- PMID: 21756339
- PMCID: PMC3152543
- DOI: 10.1186/1471-2164-12-364
Somatic sex-specific transcriptome differences in Drosophila revealed by whole transcriptome sequencing
Abstract
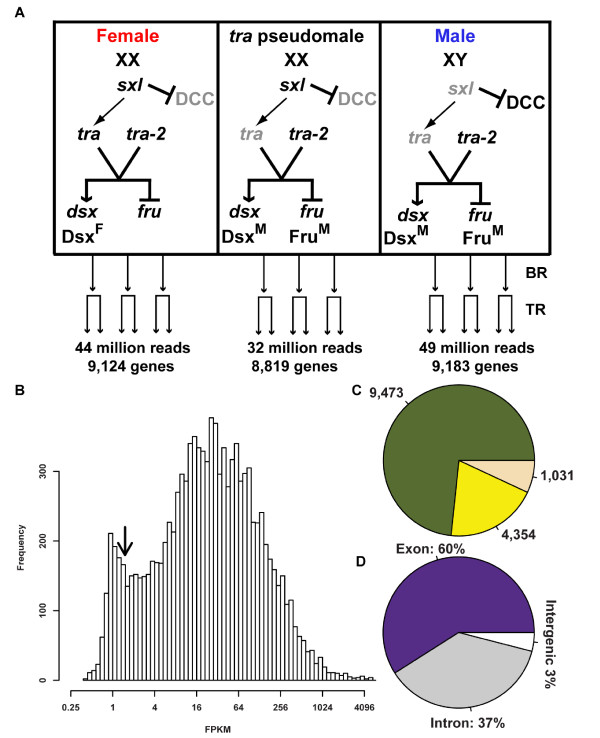
Background: Understanding animal development and physiology at a molecular-biological level has been advanced by the ability to determine at high resolution the repertoire of mRNA molecules by whole transcriptome resequencing. This includes the ability to detect and quantify rare abundance transcripts and isoform-specific mRNA variants produced from a gene.The sex hierarchy consists of a pre-mRNA splicing cascade that directs the production of sex-specific transcription factors that specify nearly all sexual dimorphism. We have used deep RNA sequencing to gain insight into how the Drosophila sex hierarchy generates somatic sex differences, by examining gene and transcript isoform expression differences between the sexes in adult head tissues.
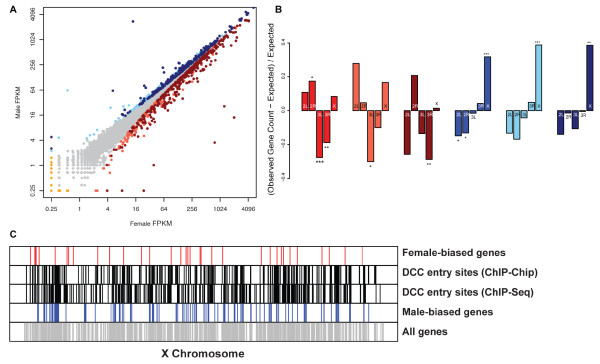
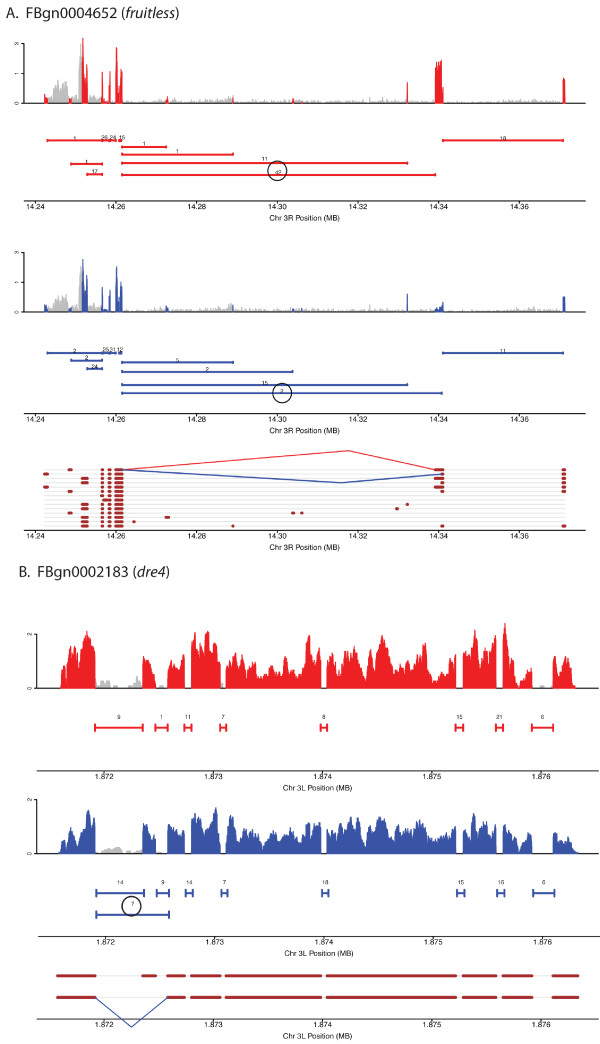
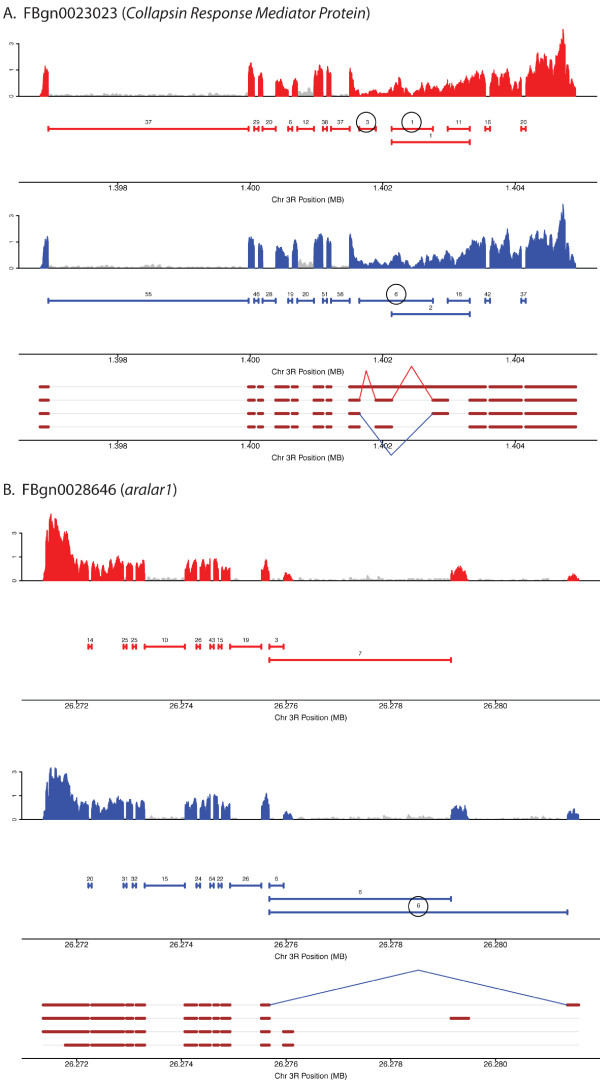
Results: Here we find 1,381 genes that differ in overall expression levels and 1,370 isoform-specific transcripts that differ between males and females. Additionally, we find 512 genes not regulated downstream of transformer that are significantly more highly expressed in males than females. These 512 genes are enriched on the × chromosome and reside adjacent to dosage compensation complex entry sites, which taken together suggests that their residence on the × chromosome might be sufficient to confer male-biased expression. There are no transcription unit structural features, from a set of features, that are robustly significantly different in the genes with significant sex differences in the ratio of isoform-specific transcripts, as compared to random isoform-specific transcripts, suggesting that there is no single molecular mechanism that generates isoform-specific transcript differences between the sexes, even though the sex hierarchy is known to include three pre-mRNA splicing factors.
Conclusions: We identify thousands of genes that show sex-specific differences in overall gene expression levels, and identify hundreds of additional genes that have differences in the abundance of isoform-specific transcripts. No transcription unit structural feature was robustly enriched in the sex-differentially expressed transcript isoforms. Additionally, we found that many genes with male-biased expression were enriched on the × chromosome and reside adjacent to dosage compensation entry sites, suggesting that differences in sex chromosome composition contributes to dimorphism in gene expression. Taken together, this study provides new insight into the molecular underpinnings of sexual differentiation.
Figures

References
-
- Consortium. Genome sequence of the nematode C. elegans: a platform for investigating biology. Science. 1998;282(5396):2012–2018. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
