

Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012
- PMID: 21760595
- PMCID: PMC3252826
- DOI: 10.1038/cdd.2011.96
Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012
Abstract
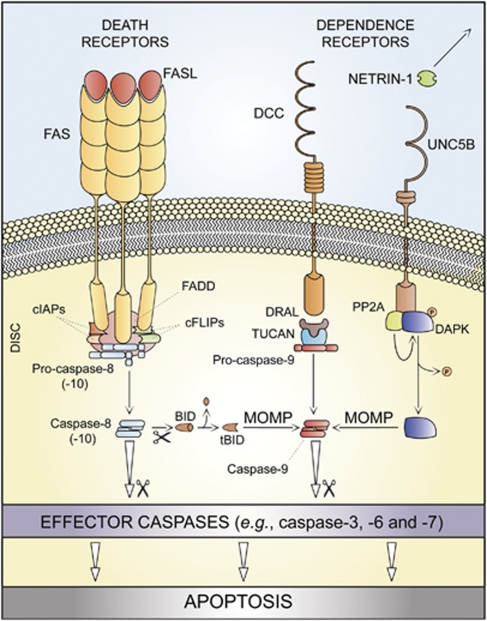
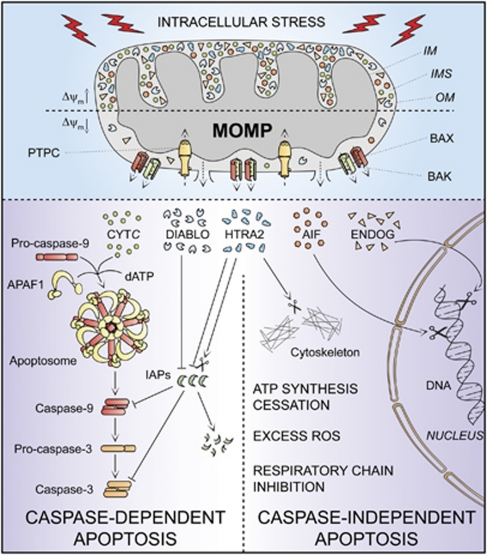
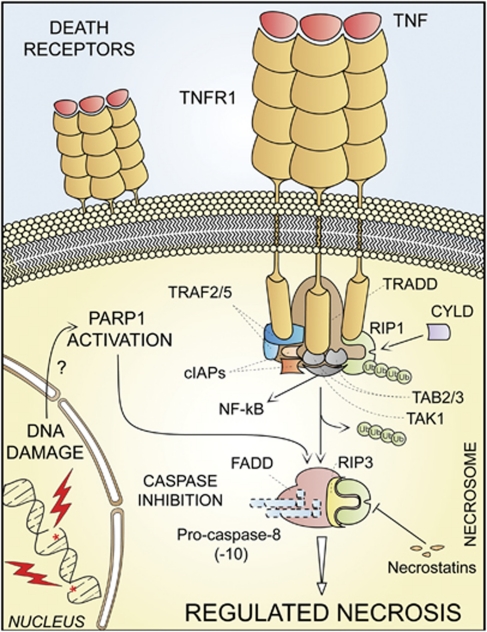
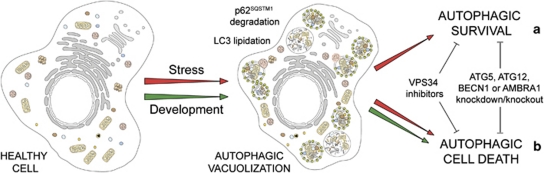
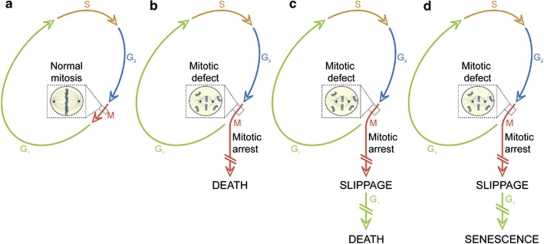
In 2009, the Nomenclature Committee on Cell Death (NCCD) proposed a set of recommendations for the definition of distinct cell death morphologies and for the appropriate use of cell death-related terminology, including 'apoptosis', 'necrosis' and 'mitotic catastrophe'. In view of the substantial progress in the biochemical and genetic exploration of cell death, time has come to switch from morphological to molecular definitions of cell death modalities. Here we propose a functional classification of cell death subroutines that applies to both in vitro and in vivo settings and includes extrinsic apoptosis, caspase-dependent or -independent intrinsic apoptosis, regulated necrosis, autophagic cell death and mitotic catastrophe. Moreover, we discuss the utility of expressions indicating additional cell death modalities. On the basis of the new, revised NCCD classification, cell death subroutines are defined by a series of precise, measurable biochemical features.
Figures
References
-
- Lockshin RA, Williams CM. Programmed cell death – II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol. 1964;10:643–649.
-
- Kerr JF. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J Pathol Bacteriol. 1965;90:419–435. - PubMed
-
- Lockshin RA, Williams CM. Programmed cell death – I. Cytology of degeneration in the intersegmental muscles of the pernyi silkmoth. J Insect Physiol. 1965;11:123–133. - PubMed
-
- Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. - PubMed
-
- Galluzzi L, Maiuri MC, Vitale I, Zischka H, Castedo M, Zitvogel L, et al. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007;14:1237–1243. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
