

Endoplasmic reticulum stress or mutation of an EF-hand Ca(2+)-binding domain directs the FKBP65 rotamase to an ERAD-based proteolysis
- PMID: 21761186
- PMCID: PMC3220392
- DOI: 10.1007/s12192-011-0270-x
Endoplasmic reticulum stress or mutation of an EF-hand Ca(2+)-binding domain directs the FKBP65 rotamase to an ERAD-based proteolysis
Abstract
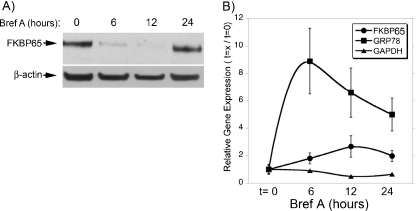
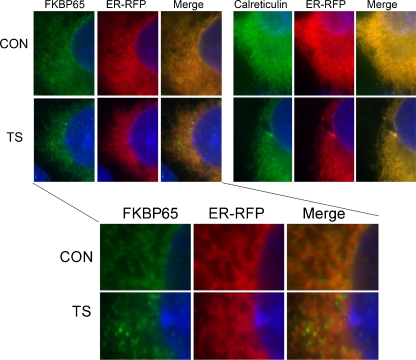
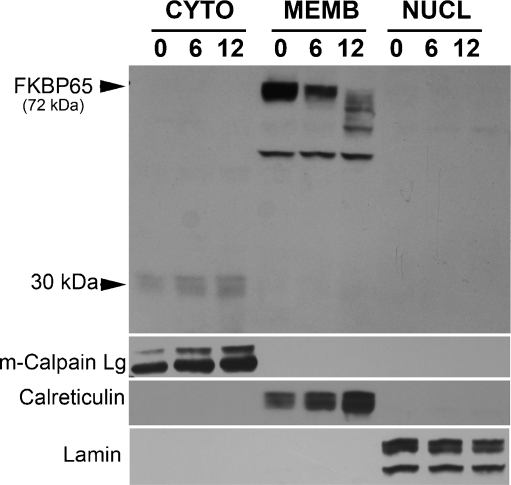
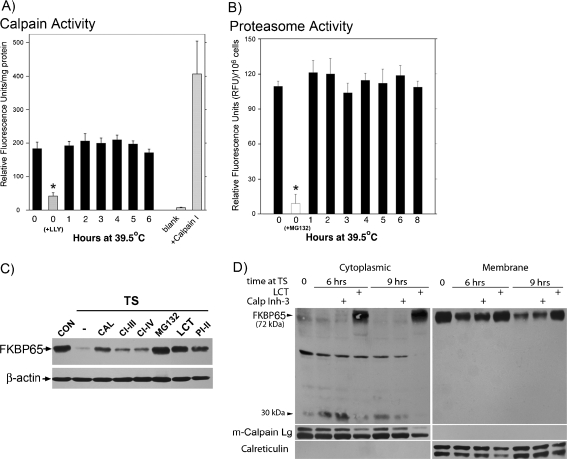
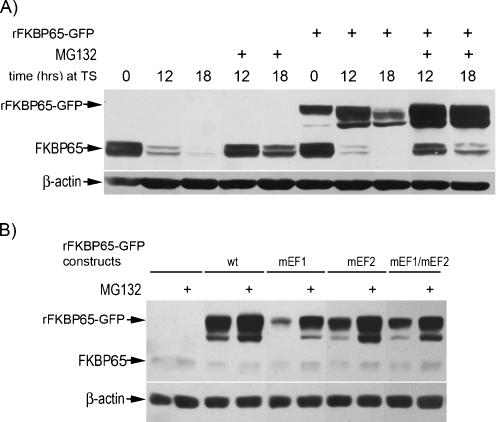
FKBP65 is an endoplasmic reticulum (ER)-localized chaperone and rotamase, with cargo proteins that include tropoelastin and collagen. In humans, mutations in FKBP65 have recently been shown to cause a form of osteogenesis imperfecta (OI), a brittle bone disease resulting from deficient secretion of mature type I collagen. In this work, we describe the rapid proteolysis of FKBP65 in response to ER stress signals that activate the release of ER Ca(2+) stores. A large-scale screen for stress-induced cellular changes revealed FKBP65 proteins to decrease within 6-12 h of stress activation. Inhibiting IP(3)R-mediated ER Ca(2+) release blocked this response. No other ER-localized chaperone and folding mediators assessed in the study displayed this phenomenon, indicating that this rapid proteolysis of folding mediator is distinctive. Imaging and cellular fractionation confirmed the localization of FKBP65 (72 kDa glycoprotein) to the ER of untreated cells, a rapid decrease in protein levels following ER stress, and the corresponding appearance of a 30-kDa fragment in the cytosol. Inhibition of the proteasome during ER stress revealed an accumulation of FKBP65 in the cytosol, consistent with retrotranslocation and a proteasome-based proteolysis. To assess the role of Ca(2+)-binding EF-hand domains in FKBP65 stability, a recombinant FKBP65-GFP construct was engineered to ablate Ca(2+) binding at each of two EF-hand domains. Cells transfected with the wild-type construct displayed ER localization of the FKBP65-GFP protein and a proteasome-dependent proteolysis in response to ER stress. Recombinant FKBP65-GFP carrying a defect in the EF1 Ca(2+)-binding domain displayed diminished protein in the ER when compared to wild-type FKBP65-GFP. Proteasome inhibition restored mutant protein to levels similar to that of the wild-type FKBP65-GFP. A similar mutation in EF2 did not confer FKBP65 proteolysis. This work supports a model in which stress-induced changes in ER Ca(2+) stores induce the rapid proteolysis of FKBP65, a chaperone and folding mediator of collagen and tropoelastin. The destruction of this protein may identify a cellular strategy for replacement of protein folding machinery following ER stress. The implications for stress-induced changes in the handling of aggregate-prone proteins in the ER-Golgi secretory pathway are discussed. This work was supported by grants from the National Institutes of Health (R15GM065139) and the National Science Foundation (DBI-0452587).
Figures


References
-
- Brewster JL, Linseman DA, Bouchard RJ, Loucks FA, Precht TA, Esch EA, Heidenreich KA. Endoplasmic reticulum stress and trophic factor withdrawal activate distinct signaling cascades that induce glycogen synthase kinase 3beta and a caspase-9-dependent apoptosis in cerebellar granule neurons. Mol Cell Neurosci. 2006;32:242–253. doi: 10.1016/j.mcn.2006.04.006. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
