

2-Methoxyestradiol inhibits progesterone-dependent tissue factor expression and activity in breast cancer cells
- PMID: 21761355
- PMCID: PMC10358164
- DOI: 10.1007/s12672-010-0019-5
2-Methoxyestradiol inhibits progesterone-dependent tissue factor expression and activity in breast cancer cells
Abstract
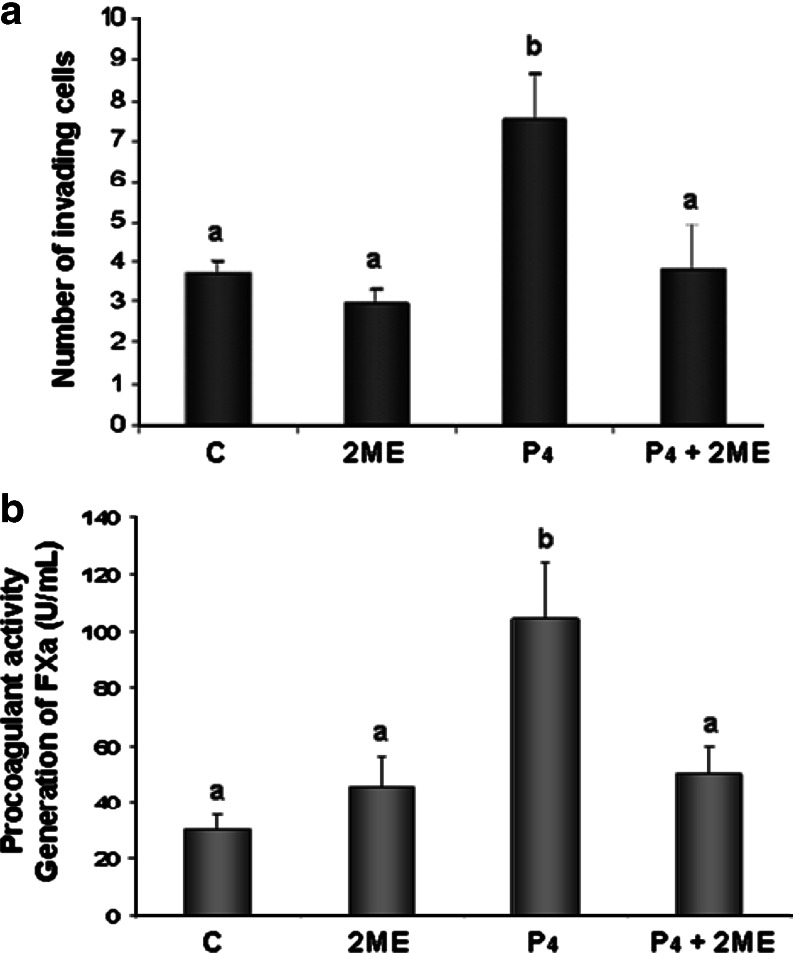
2-Methoxyestradiol (2ME) is an endogenous metabolite of 17β-estradiol with antiangiogenic and antitumor properties, although its mechanisms of action remain unclear. Progestins in hormone replacement therapy increase the risk of breast cancer. Progesterone also enhances the procoagulant activity and invasive potential of progesterone receptor (PR)-positive breast cancer cell lines, an effect largely mediated by induction of tissue factor (TF), the cellular activator of the coagulation cascade. Here we show that 2ME abrogates the induction TF expression in progesterone-treated breast cancer cells via a mechanism that does not involve the estrogen receptor. Instead, we demonstrate that by selectively antagonizing ERK1/2 signaling in breast cancer cells, 2ME limits the transactivation potential of ligand-bound PR and inhibits the expression of endogenous progesterone targets, such as TF and signal transducer and activator of transcription 5. We further demonstrate that 2ME can alter the phosphorylation status of PR. Thus, 2ME prevents progesterone-dependent increase in breast cancer cell invasiveness and procoagulant activity by uncoupling PR from the ERK1/2 signal transduction pathway.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures







References
-
- Muti P, Bradlow HL, Micheli A, Krogh V, Freudenheim JL, Schünemann HJ, Stanulla M, Yang J, Sepkovic DW, Trevisan M, Berrino F. Estrogen metabolism and risk of breast cancer: a prospective study of the 2:16alpha-hydroxyestrone ratio in premenopausal and postmenopausal women. Epidemiology. 2000;11(6):635–640. doi: 10.1097/00001648-200011000-00004. - DOI - PubMed
-
- Dahut WL, Lakhani NJ, Gulley JL, Arlen PM, Kohn EC, Kotz H, McNally D, Parr A, Nguyen D, Yang SX, Steinberg SM, Venitz J, Sparreboom A, Figg WD. Phase I clinical trial of oral 2-methoxyestradiol, an antiangiogenic and apoptotic agent, in patients with solid tumors. Cancer Biol Ther. 2006;5(1):22–27. doi: 10.4161/cbt.5.1.2349. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
