

Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis
- PMID: 21763485
- PMCID: PMC3135807
- DOI: 10.1016/j.ajhg.2011.06.002
Recessive mutations in KCNJ13, encoding an inwardly rectifying potassium channel subunit, cause leber congenital amaurosis
Abstract
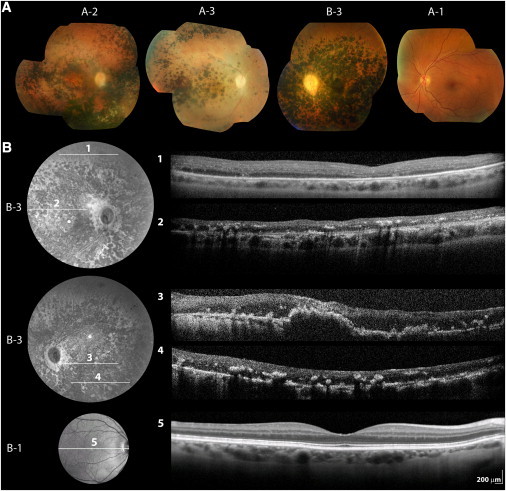
Inherited retinal degenerations, including retinitis pigmentosa (RP) and Leber congenital amaurosis (LCA), comprise a group of disorders showing high genetic and allelic heterogeneity. The determination of a full catalog of genes that can, when mutated, cause human retinal disease is a powerful means to understand the molecular physiology and pathology of the human retina. As more genes are found, remaining ones are likely to be rarer and/or unexpected candidates. Here, we identify a family in which all known RP/LCA-related genes are unlikely to be associated with their disorder. A combination of homozygosity mapping and exome sequencing identifies a homozygous nonsense mutation, c.496C>T (p.Arg166X), in a gene, KCNJ13, encoding a potassium channel subunit Kir7.1. A screen of a further 333 unrelated individuals with recessive retinal degeneration identified an additional proband, homozygous for a missense mutation, c.722T>C (p.Leu241Pro), in the same gene. The three affected members of the two families have been diagnosed with LCA. All have a distinct and unusual retinal appearance and a similar early onset of visual loss, suggesting both impaired retinal development and progressive retinal degeneration, involving both rod and cone pathways. Examination of heterozygotes revealed no ocular disease. This finding implicates Kir7.1 as having an important role in human retinal development and maintenance. This disorder adds to a small diverse group of diseases consequent upon loss or reduced function of inwardly rectifying potassium channels affecting various organs. The distinct retinal phenotype that results from biallelic mutations in KCNJ13 should facilitate the molecular diagnosis in further families.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures


References
-
- Wright A.F., Chakarova C.F., Abd El-Aziz M.M., Bhattacharya S.S. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat. Rev. Genet. 2010;11:273–284. - PubMed
-
- den Hollander A.I., Roepman R., Koenekoop R.K., Cremers F.P.M. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008;27:391–419. - PubMed
-
- Lander E.S., Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
