

Genomic analysis of hepatitis B virus reveals antigen state and genotype as sources of evolutionary rate variation
- PMID: 21765983
- PMCID: PMC3136878
- DOI: 10.3390/v3020083
Genomic analysis of hepatitis B virus reveals antigen state and genotype as sources of evolutionary rate variation
Abstract
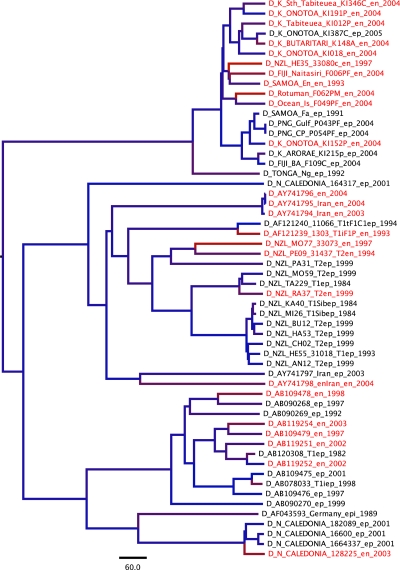
Hepatitis B virus (HBV) genomes are small, semi-double-stranded DNA circular genomes that contain alternating overlapping reading frames and replicate through an RNA intermediary phase. This complex biology has presented a challenge to estimating an evolutionary rate for HBV, leading to difficulties resolving the evolutionary and epidemiological history of the virus. Here, we re-examine rates of HBV evolution using a novel data set of 112 within-host, transmission history (pedigree) and among-host genomes isolated over 20 years from the indigenous peoples of the South Pacific, combined with 313 previously published HBV genomes. We employ Bayesian phylogenetic approaches to examine several potential causes and consequences of evolutionary rate variation in HBV. Our results reveal rate variation both between genotypes and across the genome, as well as strikingly slower rates when genomes are sampled in the Hepatitis B e antigen positive state, compared to the e antigen negative state. This Hepatitis B e antigen rate variation was found to be largely attributable to changes during the course of infection in the preCore and Core genes and their regulatory elements.
Keywords: Bayesian phylogenetics; hepatitis B virus; molecular clock.
Figures
References
-
- Drummond AJ, Pybus OG, Rambaut A, Forsberg R, Rodrigo AG. Measurably evolving populations. Trends Ecol. Evol. 2003;18:481–488.
-
- Drummond A, Pybus OG, Rambaut A. Inference of viral evolutionary rates from molecular sequences. Adv. Parasitol. 2003;54:331–358. - PubMed
-
- Nakano T, Lu L, He Y, Fu Y, Robertson BH, Pybus OG. Population genetic history of hepatitis C virus 1b infection in China. J. Gen. Virol. 2006;87:73–82. - PubMed
-
- Rodrigo AG, Goode M, Forsberg R, Ross HA, Drummond A. Inferring evolutionary rates using serially sampled sequences from several populations. Mol. Biol. Evol. 2003;20:2010–2018. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources