

Proteome-wide evidence for enhanced positive Darwinian selection within intrinsically disordered regions in proteins
- PMID: 21771306
- PMCID: PMC3218827
- DOI: 10.1186/gb-2011-12-7-r65
Proteome-wide evidence for enhanced positive Darwinian selection within intrinsically disordered regions in proteins
Abstract
Background: Understanding the adaptive changes that alter the function of proteins during evolution is an important question for biology and medicine. The increasing number of completely sequenced genomes from closely related organisms, as well as individuals within species, facilitates systematic detection of recent selection events by means of comparative genomics.
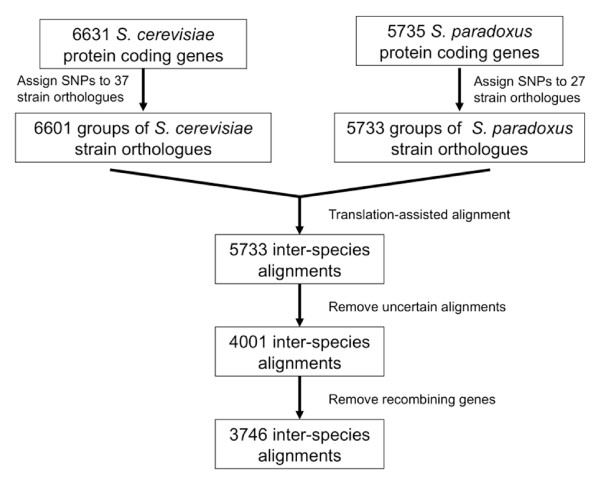
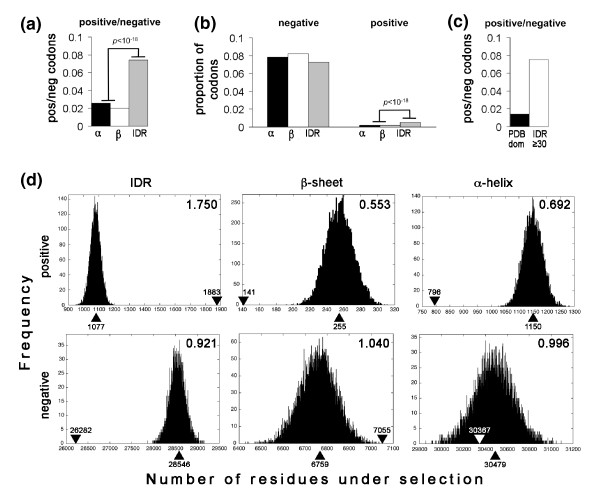
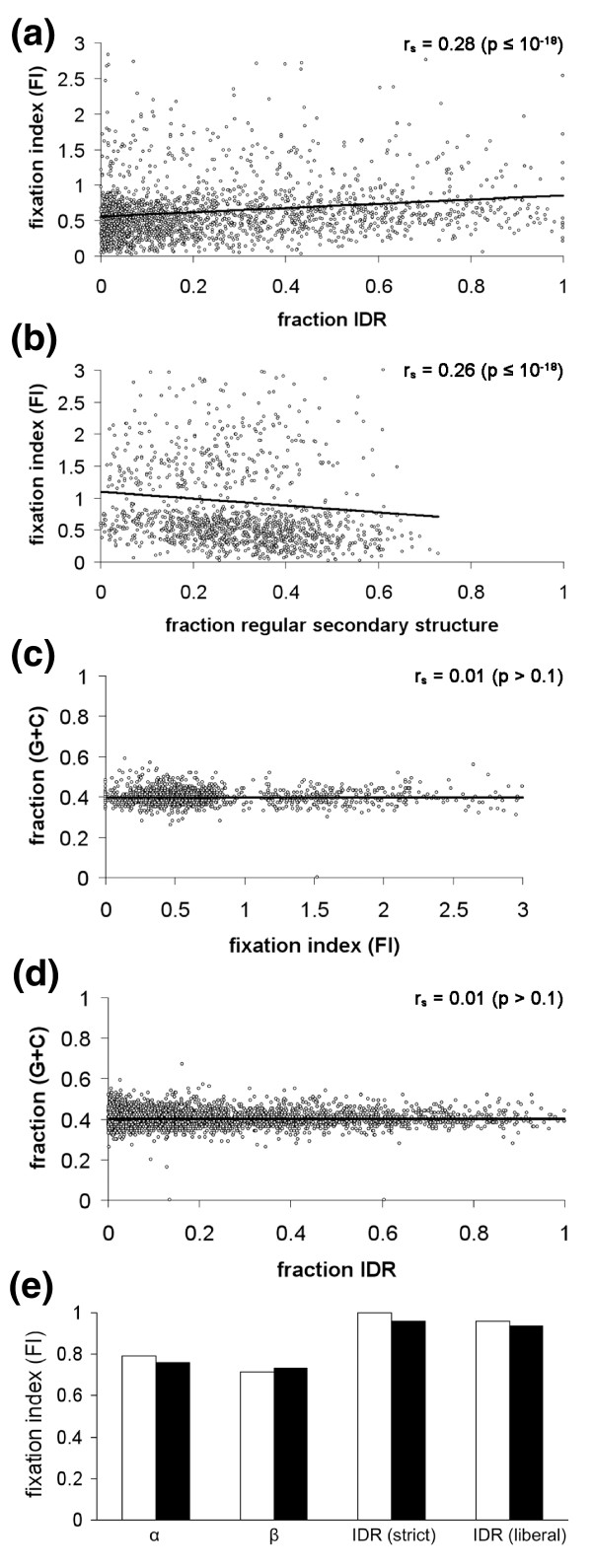
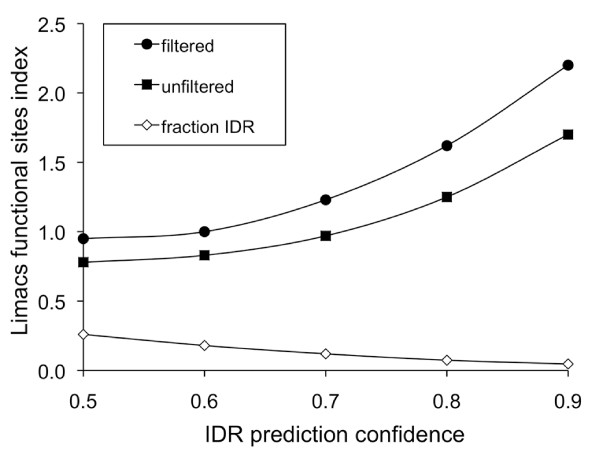
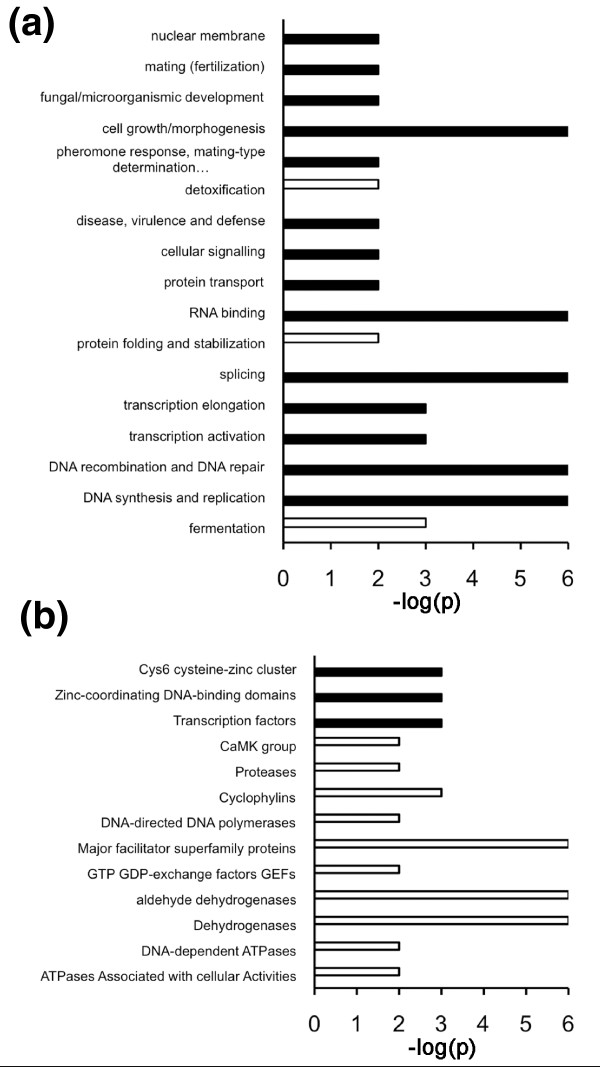
Results: We have used genome-wide strain-specific single nucleotide polymorphism data from 64 strains of budding yeast (Saccharomyces cerevisiae or Saccharomyces paradoxus) to determine whether adaptive positive selection is correlated with protein regions showing propensity for different classes of structure conformation. Data from phylogenetic and population genetic analysis of 3,746 gene alignments consistently shows a significantly higher degree of positive Darwinian selection in intrinsically disordered regions of proteins compared to regions of alpha helix, beta sheet or tertiary structure. Evidence of positive selection is significantly enriched in classes of proteins whose functions and molecular mechanisms can be coupled to adaptive processes and these classes tend to have a higher average content of intrinsically unstructured protein regions.
Conclusions: We suggest that intrinsically disordered protein regions may be important for the production and maintenance of genetic variation with adaptive potential and that they may thus be of central significance for the evolvability of the organism or cell in which they occur.
Figures

References
-
- Sabeti P, Reich D, Higgins J, Levine H, Richter D, Schaffner S, Gabriel S, Platko J, Patterson N, McDonald G, Ackerman H, Campbell S, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander E. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
