

Accurate and comprehensive sequencing of personal genomes
- PMID: 21771779
- PMCID: PMC3166834
- DOI: 10.1101/gr.123638.111
Accurate and comprehensive sequencing of personal genomes
Abstract
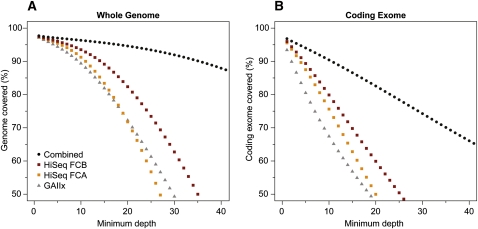
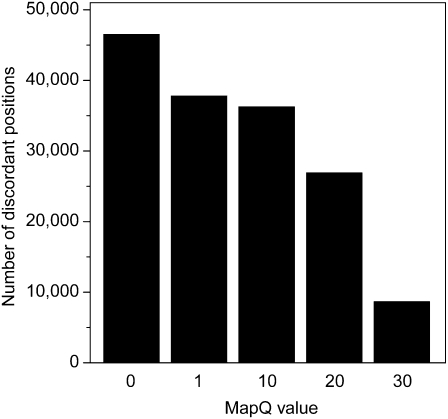
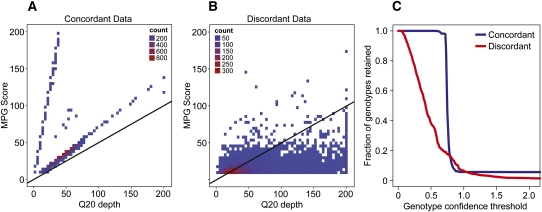
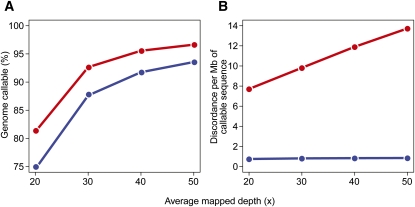
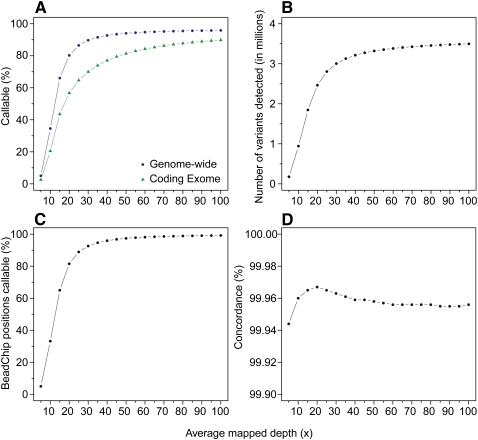
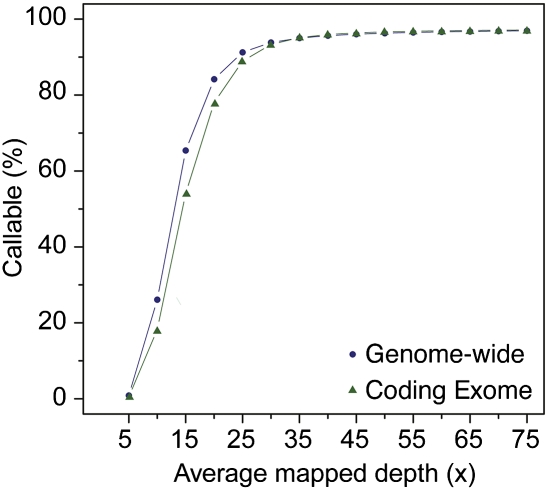
As whole-genome sequencing becomes commoditized and we begin to sequence and analyze personal genomes for clinical and diagnostic purposes, it is necessary to understand what constitutes a complete sequencing experiment for determining genotypes and detecting single-nucleotide variants. Here, we show that the current recommendation of ∼30× coverage is not adequate to produce genotype calls across a large fraction of the genome with acceptably low error rates. Our results are based on analyses of a clinical sample sequenced on two related Illumina platforms, GAII(x) and HiSeq 2000, to a very high depth (126×). We used these data to establish genotype-calling filters that dramatically increase accuracy. We also empirically determined how the callable portion of the genome varies as a function of the amount of sequence data used. These results help provide a "sequencing guide" for future whole-genome sequencing decisions and metrics by which coverage statistics should be reported.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources