

A dynamic model of proteome changes reveals new roles for transcript alteration in yeast
- PMID: 21772262
- PMCID: PMC3159980
- DOI: 10.1038/msb.2011.48
A dynamic model of proteome changes reveals new roles for transcript alteration in yeast
Abstract
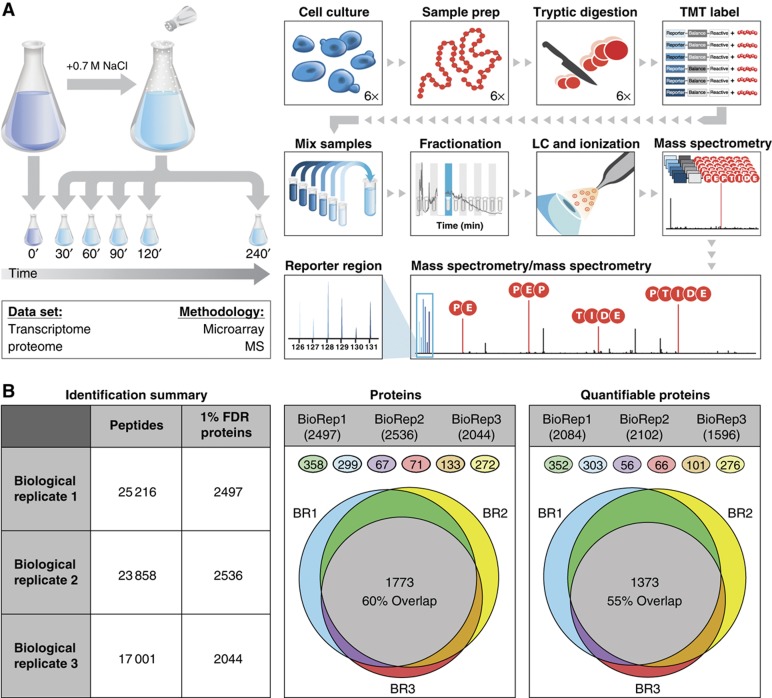
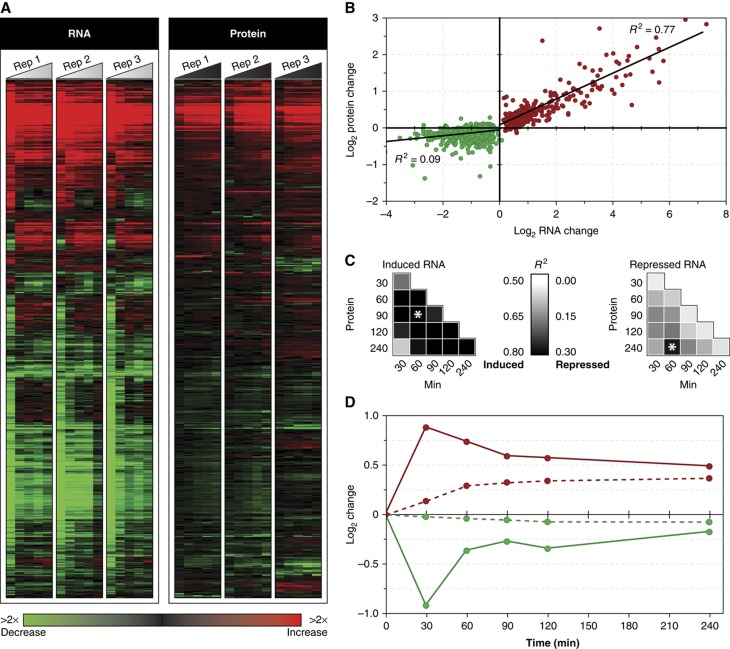
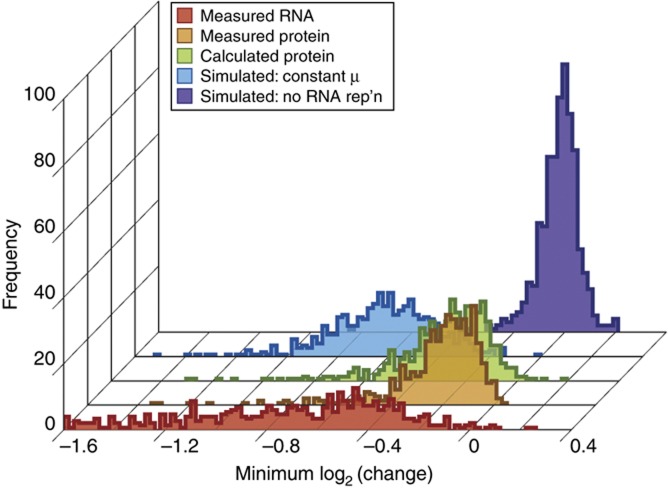
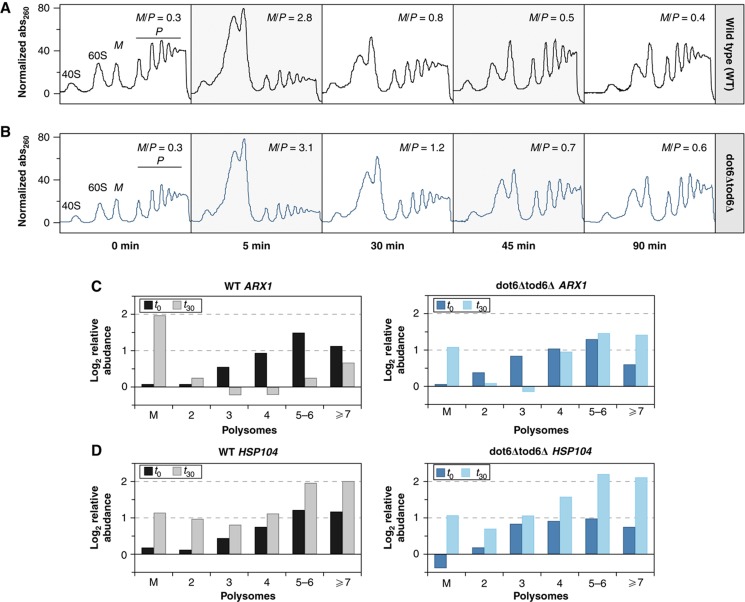
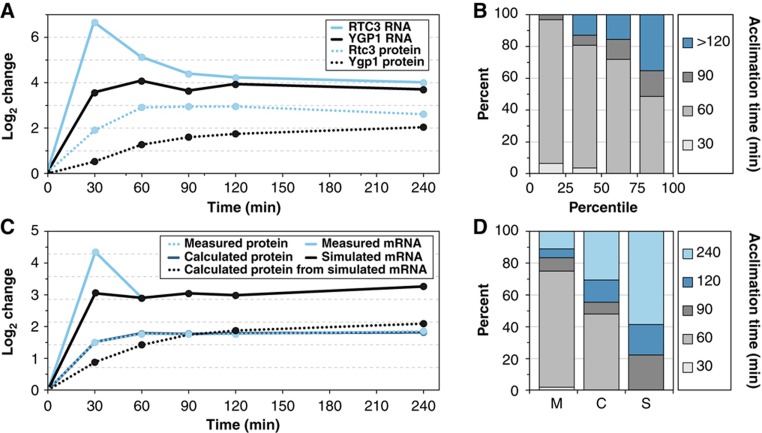
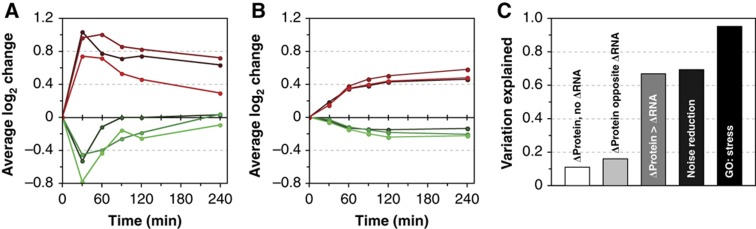
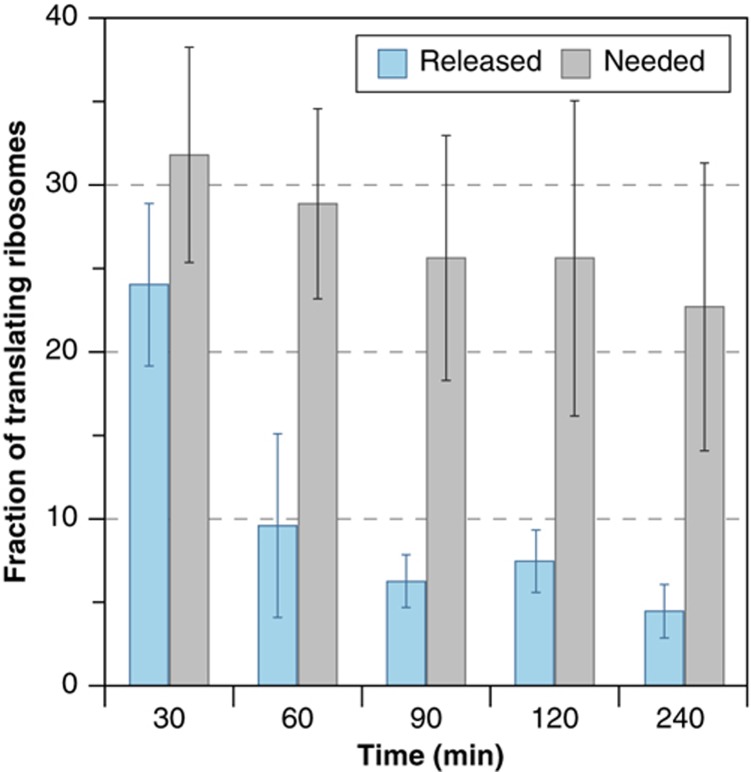
The transcriptome and proteome change dynamically as cells respond to environmental stress; however, prior proteomic studies reported poor correlation between mRNA and protein, rendering their relationships unclear. To address this, we combined high mass accuracy mass spectrometry with isobaric tagging to quantify dynamic changes in ~2500 Saccharomyces cerevisiae proteins, in biological triplicate and with paired mRNA samples, as cells acclimated to high osmolarity. Surprisingly, while transcript induction correlated extremely well with protein increase, transcript reduction produced little to no change in the corresponding proteins. We constructed a mathematical model of dynamic protein changes and propose that the lack of protein reduction is explained by cell-division arrest, while transcript reduction supports redistribution of translational machinery. Furthermore, the transient 'burst' of mRNA induction after stress serves to accelerate change in the corresponding protein levels. We identified several classes of post-transcriptional regulation, but show that most of the variance in protein changes is explained by mRNA. Our results present a picture of the coordinated physiological responses at the levels of mRNA, protein, protein-synthetic capacity, and cellular growth.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev 8: 450–461 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
