

Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species
- PMID: 21772945
- PMCID: PMC3139040
- DOI: 10.4330/wjc.v3.i6.186
Ischemia/reperfusion injury and cardioprotective mechanisms: Role of mitochondria and reactive oxygen species
Abstract
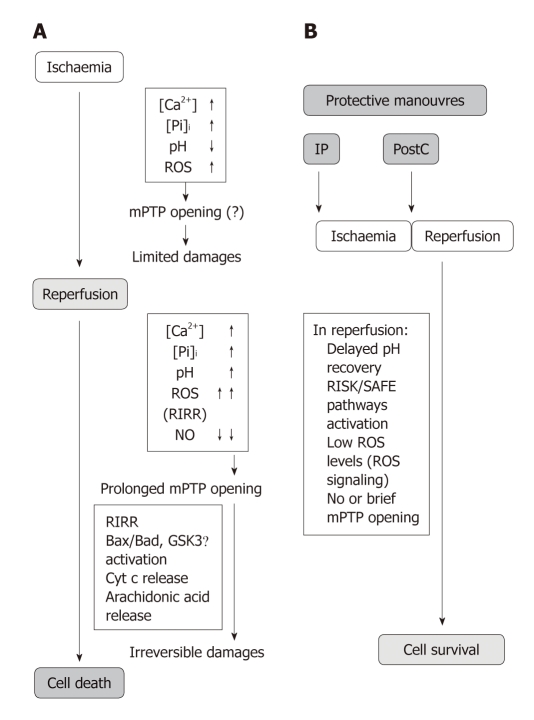
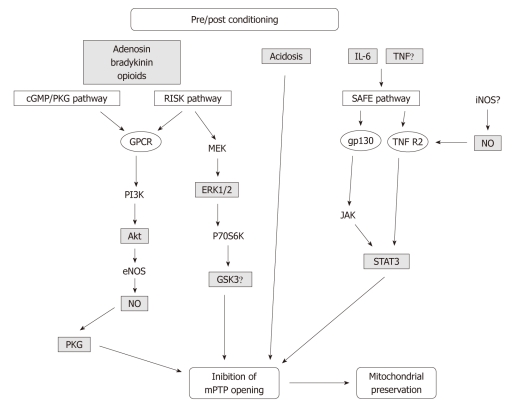
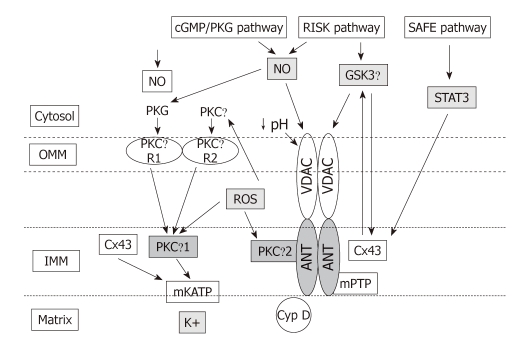
Reperfusion therapy must be applied as soon as possible to attenuate the ischemic insult of acute myocardial infarction (AMI). However reperfusion is responsible for additional myocardial damage, which likely involves opening of the mitochondrial permeability transition pore (mPTP). In reperfusion injury, mitochondrial damage is a determining factor in causing loss of cardiomyocyte function and viability. Major mechanisms of mitochondrial dysfunction include the long lasting opening of mPTPs and the oxidative stress resulting from formation of reactive oxygen species (ROS). Several signaling cardioprotective pathways are activated by stimuli such as preconditioning and postconditioning, obtained with brief intermittent ischemia or with pharmacological agents. These pathways converge on a common target, the mitochondria, to preserve their function after ischemia/reperfusion. The present review discusses the role of mitochondria in cardioprotection, especially the involvement of adenosine triphosphate-dependent potassium channels, ROS signaling, and the mPTP. Ischemic postconditioning has emerged as a new way to target the mitochondria, and to drastically reduce lethal reperfusion injury. Several clinical studies using ischemic postconditioning during angioplasty now support its protective effects, and an interesting alternative is pharmacological postconditioning. In fact ischemic postconditioning and the mPTP desensitizer, cyclosporine A, have been shown to induce comparable protection in AMI patients.
Keywords: Adenosine triphosphate-dependent potassium channels; Cardioprotection; Ischemia-reperfusion injury; Mitochondrial permeability transition pore; Reactive oxygen species.
Figures
References
-
- Keeley EC, Boura JA, Grines CL. Primary angioplasty versus intravenous thrombolytic therapy for acute myocardial infarction: a quantitative review of 23 randomised trials. Lancet. 2003;361:13–20. - PubMed
-
- Penna C, Mancardi D, Rastaldo R, Pagliaro P. Cardioprotection: a radical view Free radicals in pre and postconditioning. Biochim Biophys Acta. 2009;1787:781–793. - PubMed
-
- Pagliaro P, Moro F, Tullio F, Perrelli MG, Penna C. Cardioprotective pathways during reperfusion: focus on redox signaling and other modalities of cell signaling. Antioxid Redox Signal. 2011;14:833–850. - PubMed
-
- Vinten-Johansen J, Granfeldt A, Mykytenko J, Undyala VV, Dong Y, Przyklenk K. The multidimensional physiological responses to postconditioning. Antioxid Redox Signal. 2011;14:791–810. - PubMed
