

Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-Mediated Neurodegeneration
- PMID: 21776259
- PMCID: PMC3135156
- DOI: 10.1155/2011/405194
Protective Action of Neurotrophic Factors and Estrogen against Oxidative Stress-Mediated Neurodegeneration
Abstract
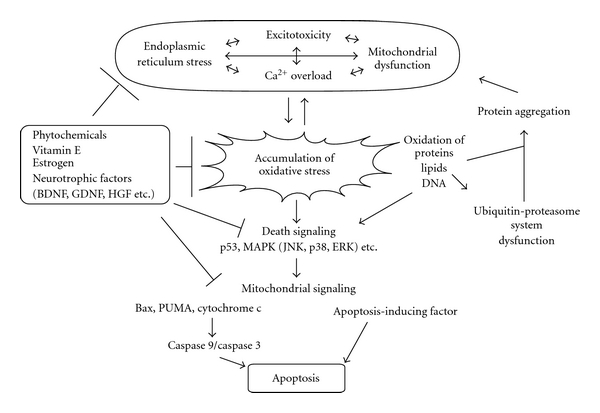
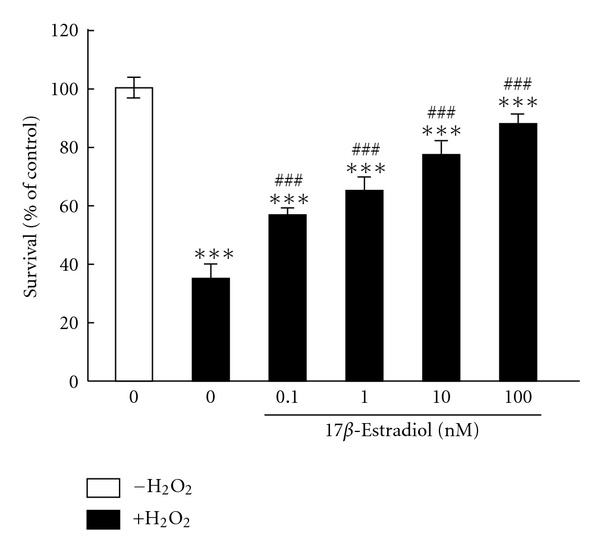
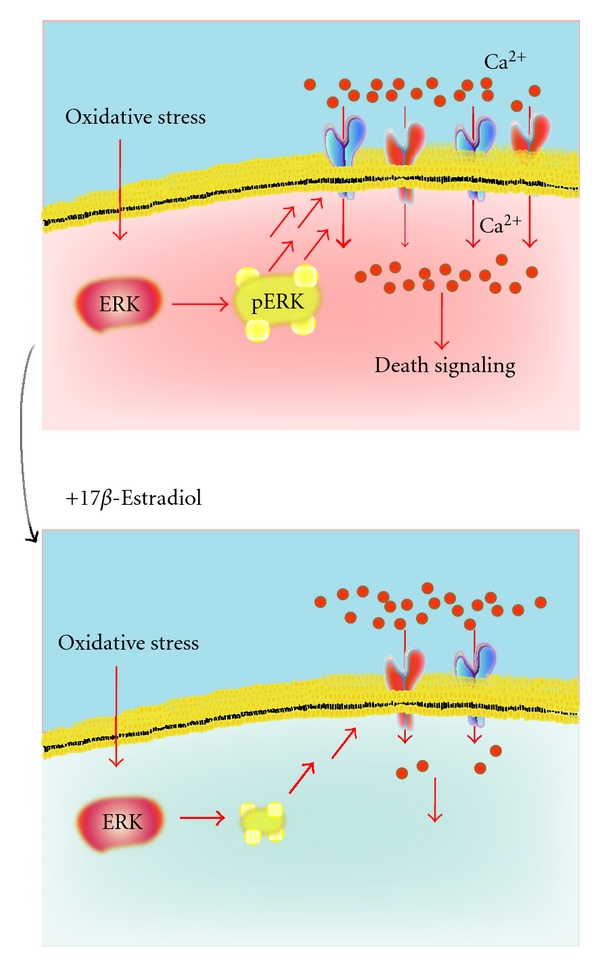
Oxidative stress is involved in the pathogenesis of neurodegenerative disorders such as Alzheimer's disease, Parkinson's disease, and Huntington's disease. Low levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) are important for maintenance of neuronal function, though elevated levels lead to neuronal cell death. A complex series of events including excitotoxicity, Ca(2+) overload, and mitochondrial dysfunction contributes to oxidative stress-mediated neurodegeneration. As expected, many antioxidants like phytochemicals and vitamins are known to reduce oxidative toxicity. Additionally, growing evidence indicates that neurotrophic factors such as brain-derived neurotrophic factor (BDNF) and estrogens significantly prevent neuronal damage caused by oxidative stress. Here, we review and discuss recent studies addressing the protective mechanisms of neurotrophic factors and estrogen within this system.
Figures
References
-
- Sakurada O, Kennedy C, Jehle J, Brown JD, Carbin GL, Sokoloff L. Measurement of local cerebral blood flow with iodo [14C] antipyrine. The American Journal of Physiology. 1978;234(1):H59–H66. - PubMed
-
- Cremer JE, Seville MP. Regional brain blood flow, blood volume, and haematocrit values in the adult rat. Journal of Cerebral Blood Flow and Metabolism. 1983;3(2):254–256. - PubMed
-
- Scremin OU, Sonnenschein RR, Rubinstein EH. Cholinergic cerebral vasodilatation: lack of involvement of cranial parasympathetic nerves. Journal of Cerebral Blood Flow and Metabolism. 1983;3(3):362–368. - PubMed
-
- Sokoloff L, Reivich M, Kennedy C, et al. The [C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. Journal of Neurochemistry. 1977;28(5):897–916. - PubMed
-
- Sokoloff L. Localization of functional activity in the central nervous system by measurement of glucose utilization with radioactive deoxyglucose. Journal of Cerebral Blood Flow and Metabolism. 1981;1(1):7–36. - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
