

Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression
- PMID: 21778339
- PMCID: PMC3172806
- DOI: 10.1182/blood-2011-01-331694
Pericytes promote endothelial cell survival through induction of autocrine VEGF-A signaling and Bcl-w expression
Abstract
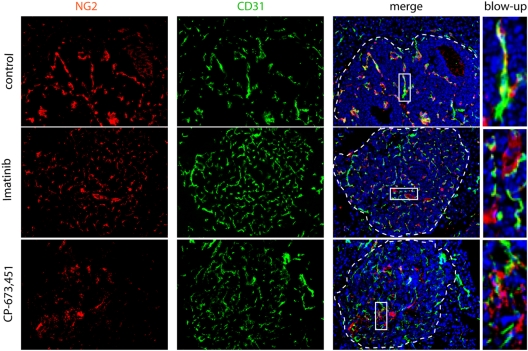
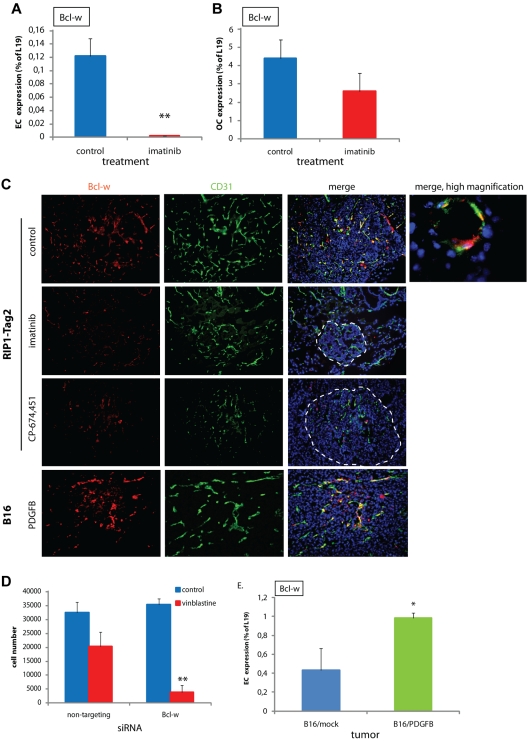
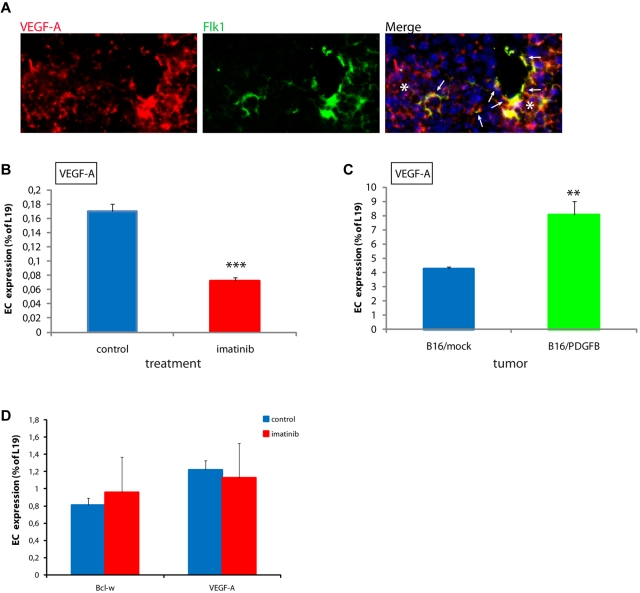
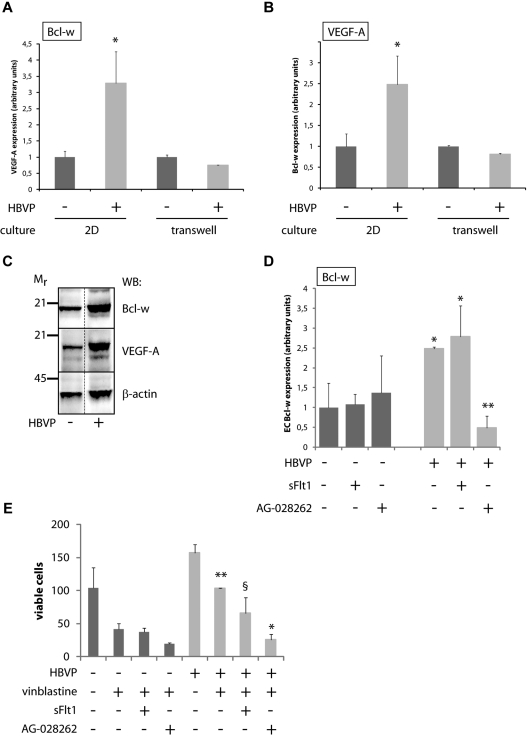
Endothelial cells (ECs) in blood vessels under formation are stabilized by the recruitment of pericytes, both in normal tissues and during angiogenesis in pathologic situations, including neoplasia. In the tumor vasculature, besides supporting the functionality of blood flow, pericytes protect ECs from antiangiogenic therapies, and have thus been implicated in clinical resistance to vascular targeting drugs. However, the molecular nature of the crosstalk between pericytes and ECs is largely unchartered. Herein, we identified pericyte-induced survival signals in ECs by isolation of vascular fragments derived from tumors that had been genetically or pharmacologically engineered to be either pericyte-rich or pericyte-poor. Pericytes induced the antiapoptotic protein Bcl-w in tumor ECs both in vivo and in vitro, thereby conveying protection from cytotoxic damage. The pericyte-dependent survival signaling in ECs was consequential to enforcement of an autocrine loop involving VEGF-A expression in ECs. Through molecular and functional studies, we delineated a signal transduction pathway in ECs downstream of integrin α(v) involving activation of NF-κB as the initiating event of the protective crosstalk from pericytes. Our elucidation of pericyte-derived pro-survival signaling in tumor ECs has potentially important implications for clinical development of antiangiogenic drugs, and suggests new therapeutic targets for rational multitargeting of cancer.
Figures



Comment in
-
Pericyte-endothelial cell interaction: a survival mechanism for the tumor vasculature.Cell Adh Migr. 2012 May-Jun;6(3):157-9. doi: 10.4161/cam.20252. Epub 2012 May 1. Cell Adh Migr. 2012. PMID: 22568989 Free PMC article. No abstract available.
References
-
- Pietras K, Ostman A. Hallmarks of cancer: interactions with the tumor stroma. Exp Cell Res. 2010;316(8):1324–1331. - PubMed
-
- Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of vegf signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299–309. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
