

Endothelin-1, the unfolded protein response, and persistent inflammation: role of pulmonary artery smooth muscle cells
- PMID: 21778413
- PMCID: PMC3262656
- DOI: 10.1165/rcmb.2010-0506OC
Endothelin-1, the unfolded protein response, and persistent inflammation: role of pulmonary artery smooth muscle cells
Abstract
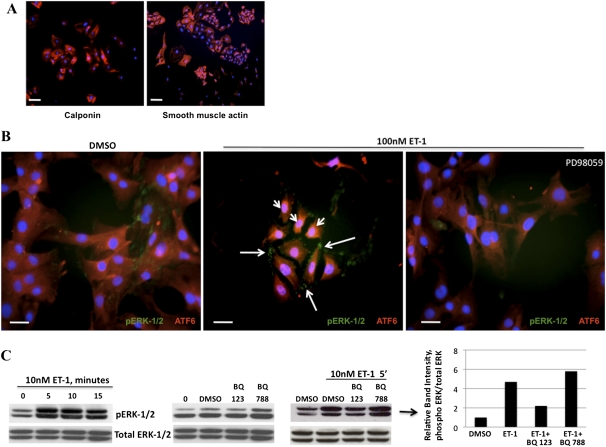
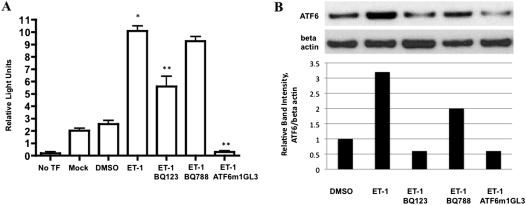
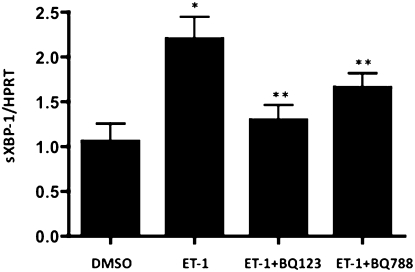
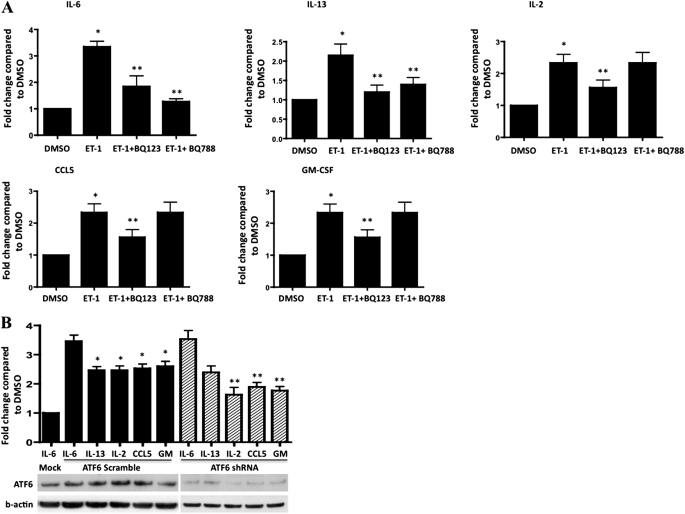
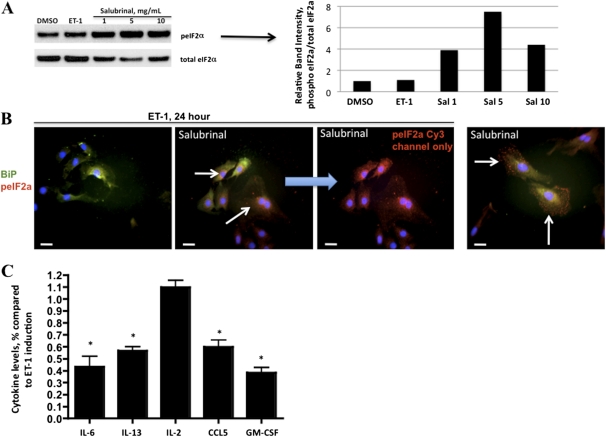
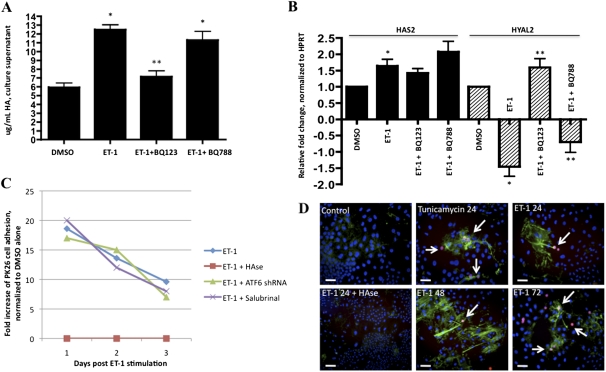
Endothelin-1 is a potent vasoactive peptide that occurs in chronically high levels in humans with pulmonary hypertension and in animal models of the disease. Recently, the unfolded protein response was implicated in a variety of diseases, including pulmonary hypertension. In addition, evidence is increasing for pathological, persistent inflammation in the pathobiology of this disease. We investigated whether endothelin-1 might engage the unfolded protein response and thus link inflammation and the production of hyaluronic acid by pulmonary artery smooth muscle cells. Using immunoblot, real-time PCR, immunofluorescence, and luciferase assays, we found that endothelin-1 induces both a transcriptional and posttranslational activation of the three major arms of the unfolded protein response. The pharmacologic blockade of endothelin A receptors, but not endothelin B receptors, attenuated the observed release, as did a pharmacologic blockade of extracellular signal-regulated kinases 1 and 2 (ERK-1/2) signaling. Using short hairpin RNA and ELISA, we observed that the release by pulmonary artery smooth muscle cells of inflammatory modulators, including hyaluronic acid, is associated with endothelin-1-induced ERK-1/2 phosphorylation and the unfolded protein response. Furthermore, the synthesis of hyaluronic acid induced by endothelin-1 is permissive for persistent THP-1 monocyte binding. These results suggest that endothelin-1, in part because it induces the unfolded protein response in pulmonary artery smooth muscle cells, triggers proinflammatory processes that likely contribute to vascular remodeling in pulmonary hypertension.
Figures

References
-
- Milan A, Magnino C, Veglio F. Echocardiographic indexes for the non-invasive evaluation of pulmonary hemodynamics. J Am Soc Echocardiogr 2010;23:225–239 - PubMed
-
- Hassoun PM, Mouthon L, Barberà JA, Eddahibi S, Flores SC, Grimminger F, Jones PL, Maitland ML, Michelakis ED, Morrell NW, et al. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol 2009;54:S10–S19 - PubMed
-
- Dorfmuller P, Humbert M, Perros F, Sanchez O, Simonneau G, Muller KM, Capron F. Fibrous remodeling of the pulmonary venous system in pulmonary arterial hypertension associated with connective tissue diseases. Hum Pathol 2007;38:893–902 - PubMed
-
- Dorfmuller P, Zarka V, Durand-Gasselin I, Monti G, Balabanian K, Garcia G, Capron F, Coulomb-Lhermine A, Marfaing-Koka A, Simonneau G, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med 2002;165:534–539 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
