

S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors
- PMID: 21779383
- PMCID: PMC3133657
- DOI: 10.1371/journal.pone.0022126
S100B and APP promote a gliocentric shift and impaired neurogenesis in Down syndrome neural progenitors
Abstract
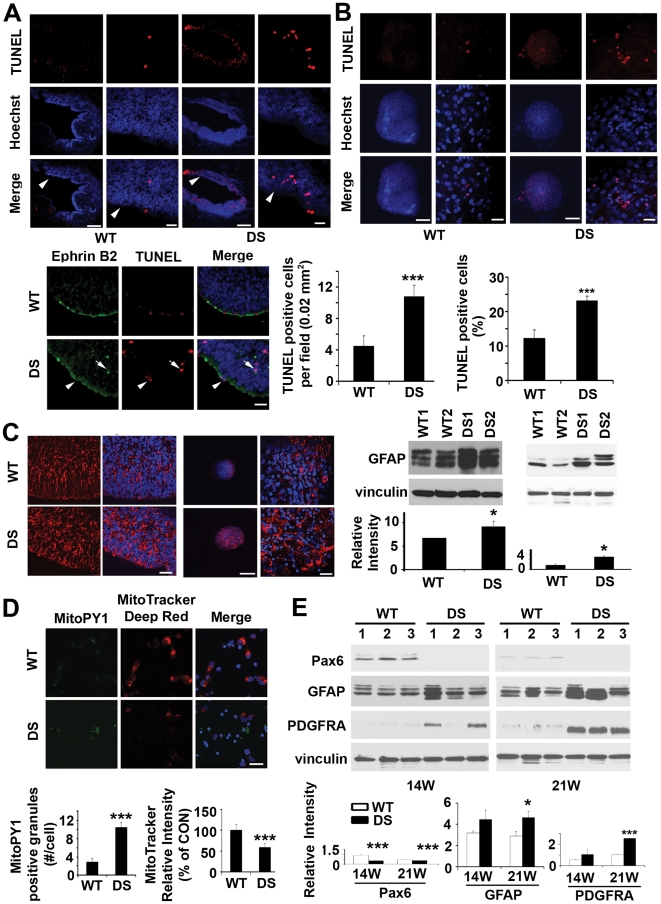
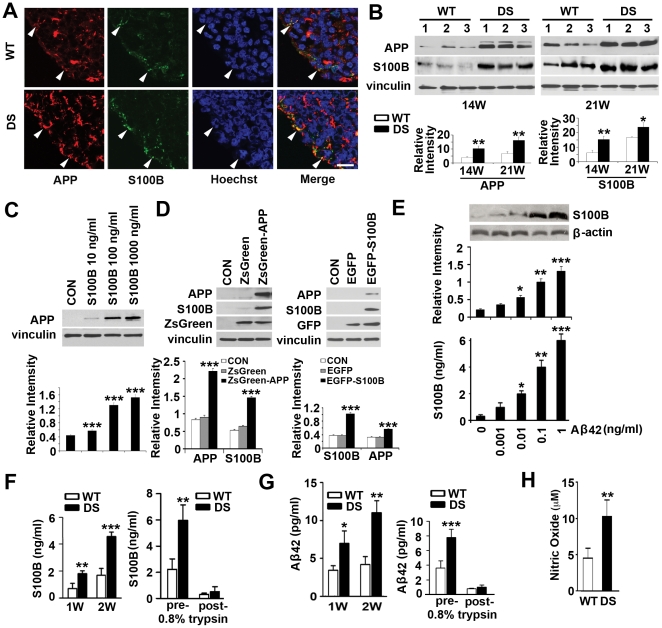
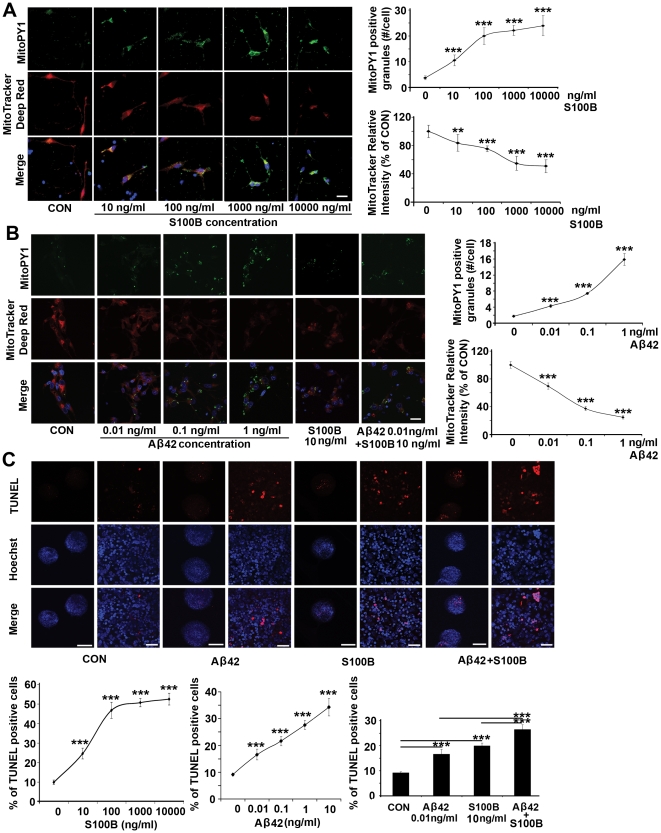
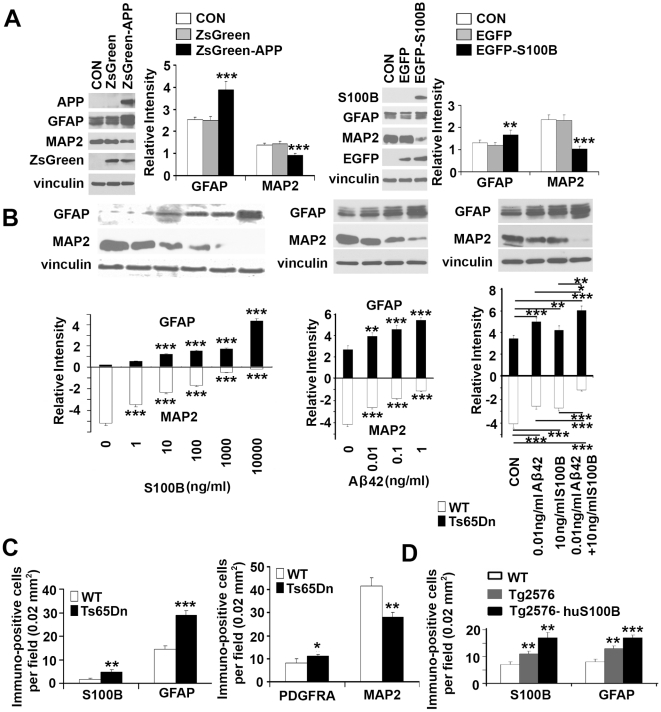
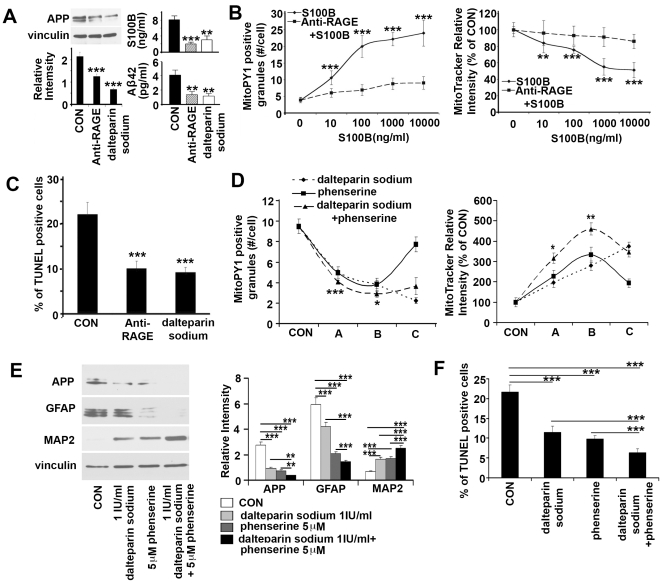
Down syndrome (DS) is a developmental disorder associated with mental retardation (MR) and early onset Alzheimer's disease (AD). These CNS phenotypes are attributed to ongoing neuronal degeneration due to constitutive overexpression of chromosome 21 (HSA21) genes. We have previously shown that HSA21 associated S100B contributes to oxidative stress and apoptosis in DS human neural progenitors (HNPs). Here we show that DS HNPs isolated from fetal frontal cortex demonstrate not only disturbances in redox states within the mitochondria and increased levels of progenitor cell death but also transition to more gliocentric progenitor phenotypes with a consequent reduction in neuronogenesis. HSA21 associated S100B and amyloid precursor protein (APP) levels are simultaneously increased within DS HNPs, their secretions are synergistically enhanced in a paracrine fashion, and overexpressions of these proteins disrupt mitochondrial membrane potentials and redox states. HNPs show greater susceptibility to these proteins as compared to neurons, leading to cell death. Ongoing inflammation through APP and S100B overexpression further promotes a gliocentric HNPs phenotype. Thus, the loss in neuronal numbers seen in DS is not merely due to increased HNPs cell death and neurodegeneration, but also a fundamental gliocentric shift in the progenitor pool that impairs neuronal production.
Conflict of interest statement
Figures
References
-
- Contestabile A, Fila T, Ceccarelli C, Bonasoni P, Bonapace L, et al. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus. 2007;17:665–678. - PubMed
-
- Busciglio J, Pelsman A, Wong C, Pigino G, Yuan M, et al. Altered metabolism of the amyloid beta precursor protein is associated with mitochondrial dysfunction in Down's syndrome. Neuron. 2002;33:677–688. - PubMed
-
- Seidl R, Bidmon B, Bajo M, Yoo PC, Cairns N, et al. Evidence for apoptosis in the fetal Down syndrome brain. J Child Neurol. 2001;16:438–442. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
