

Anti-CXCL5 therapy ameliorates IL-17-induced arthritis by decreasing joint vascularization
- PMID: 21779896
- PMCID: PMC4281892
- DOI: 10.1007/s10456-011-9227-z
Anti-CXCL5 therapy ameliorates IL-17-induced arthritis by decreasing joint vascularization
Abstract
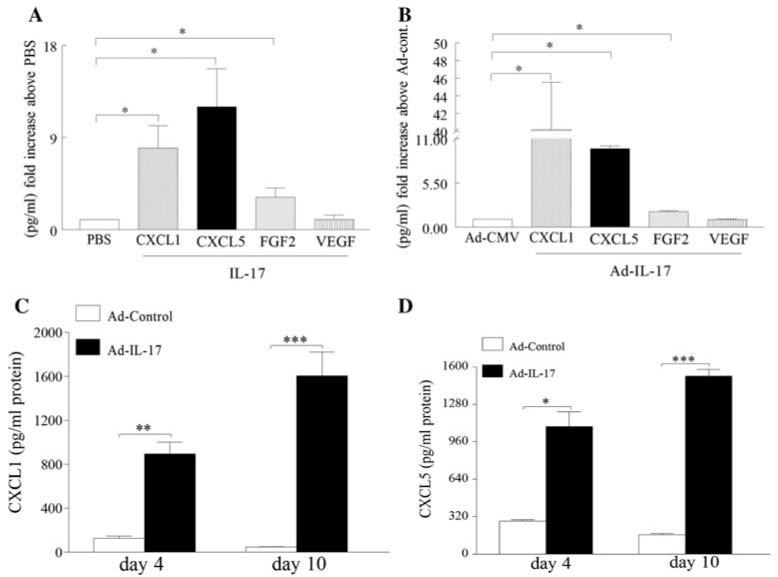
IL-17-induced joint inflammation is associated with increased angiogenesis. However, the mechanism by which IL-17 mediates angiogenesis is undefined. Therefore, the pathologic role of CXCL1 and CXCL5 was investigated in arthritis mediated by local expression of IL-17, employing a neutralizing antibody to each chemokine. Next, endothelial chemotaxis was utilized to examine whether endothelial migration was differentially mediated by CXCL1 and CXCL5. Our results demonstrate that IL-17-mediated disease activity was not affected by anti-CXCL1 treatment alone. In contrast, mice receiving anti-CXCL5 demonstrated significantly reduced clinical signs of arthritis, compared to the mice treated with IgG control. Consistently, while inflammation, synovial lining thickness, bone erosion and vascularization were markedly reduced in both the anti-CXCL5 and combination anti-CXCL1 and 5 treatment groups, mice receiving anti-CXCL1 antibody had clinical scores similar to the control group. In contrast to joint FGF2 and VEGF levels, TNF-α was significantly reduced in mice receiving anti-CXCL5 or combination of anti-CXCL1 and 5 therapies compared to the control group. We found that, like IL-17, CXCL1-induced endothelial migration is mediated through activation of PI3K. In contrast, activation of NF-κB pathway was essential for endothelial chemotaxis induced by CXCL5. Although CXCL1 and CXCL5 can differentially mediate endothelial trafficking, blockade of CXCR2 can inhibit endothelial chemotaxis mediated by either of these chemokines. These results suggest that blockade of CXCL5 can modulate IL-17-induced inflammation in part by reducing joint blood vessel formation through a non-overlapping IL-17 mechanism.
Figures






References
-
- Firestein GS. Evolving concepts of rheumatoid arthritis. Nature. 2003;423:356–361. - PubMed
-
- Lubberts E, Koenders MI, Oppers-Walgreen B, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–659. - PubMed
-
- Koenders MI, Lubberts E, van de Loo FA, et al. Interleukin-17 acts independently of TNF-alpha under arthritic conditions. J Immunol. 2006;176:6262–6269. - PubMed
-
- Lubberts E, Joosten LA, Oppers B, et al. IL-1-independent role of IL-17 in synovial inflammation and joint destruction during collagen-induced arthritis. J Immunol. 2001;167:1004–1013. - PubMed
-
- Koenders MI, Lubberts E, Oppers-Walgreen B, et al. Induction of cartilage damage by overexpression of T cell interleukin-17A in experimental arthritis in mice deficient in interleukin-1. Arthritis Rheum. 2005;52:975–983. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
