

Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity
- PMID: 21782022
- PMCID: PMC3220788
- DOI: 10.1016/j.jsbmb.2011.07.002
Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity
Abstract
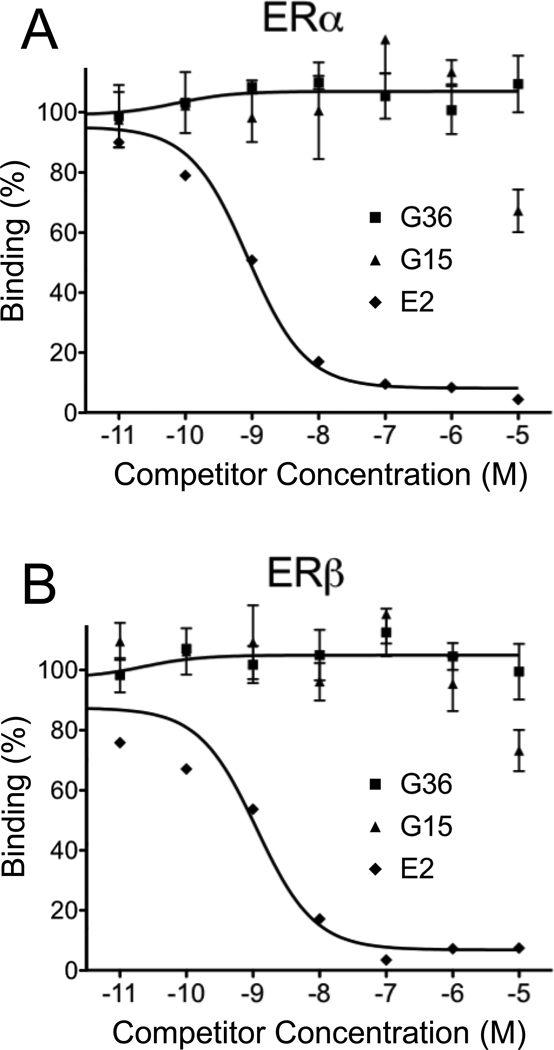
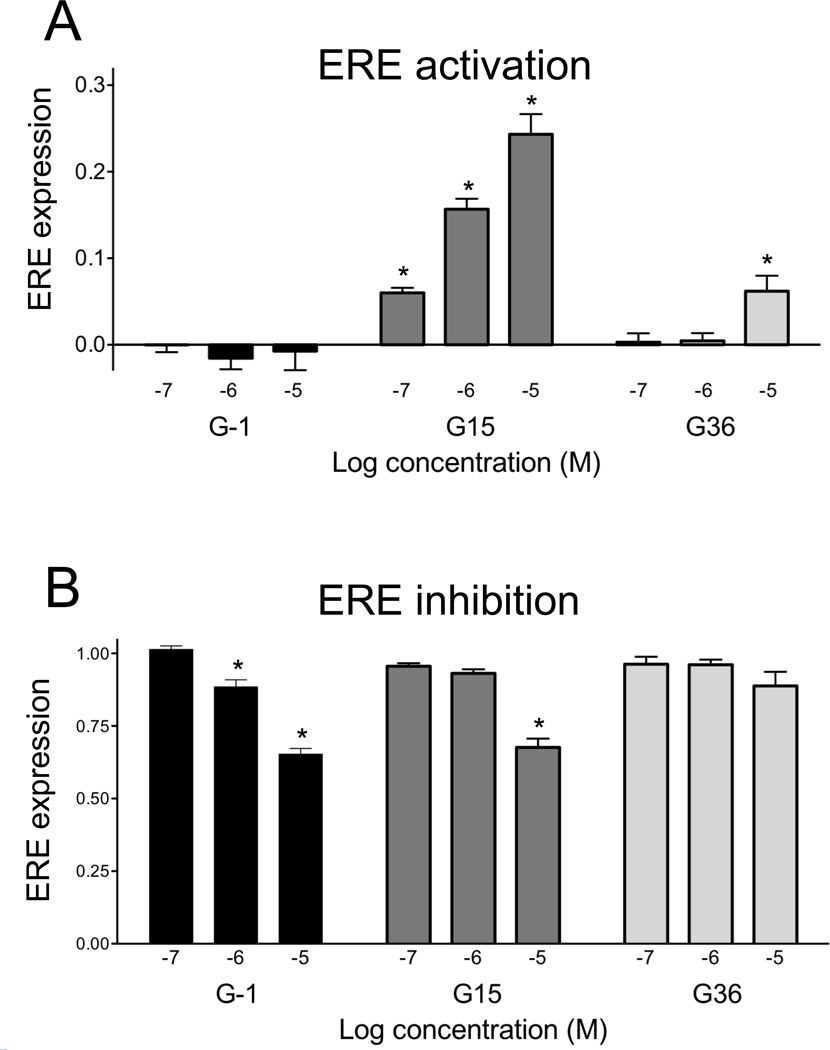
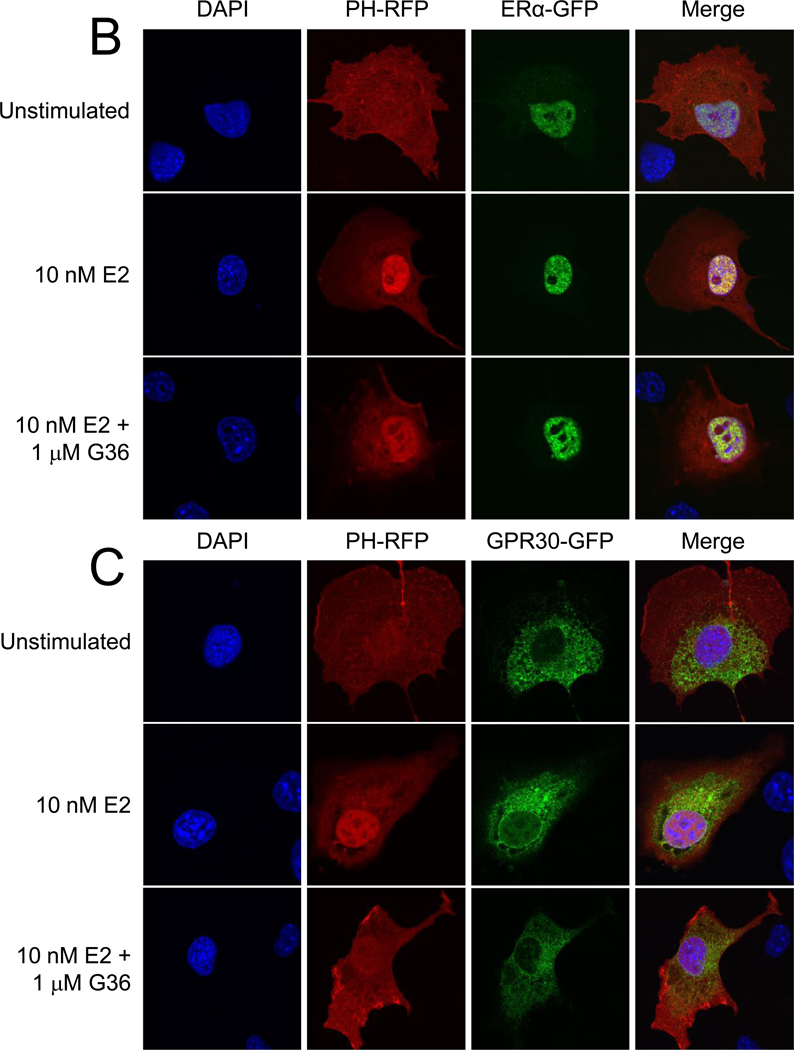
GPER/GPR30 is a seven-transmembrane G protein-coupled estrogen receptor that regulates many aspects of mammalian biology and physiology. We have previously described both a GPER-selective agonist G-1 and antagonist G15 based on a tetrahydro-3H-cyclopenta[c]quinoline scaffold. The antagonist lacks an ethanone moiety that likely forms important hydrogen bonds involved in receptor activation. Computational docking studies suggested that the lack of the ethanone substituent in G15 could minimize key steric conflicts, present in G-1, that limit binding within the ERα ligand binding pocket. In this report, we identify low-affinity cross-reactivity of the GPER antagonist G15 to the classical estrogen receptor ERα. To generate an antagonist with enhanced selectivity, we therefore synthesized an isosteric G-1 derivative, G36, containing an isopropyl moiety in place of the ethanone moiety. We demonstrate that G36 shows decreased binding and activation of ERα, while maintaining its antagonist profile towards GPER. G36 selectively inhibits estrogen-mediated activation of PI3K by GPER but not ERα. It also inhibits estrogen- and G-1-mediated calcium mobilization as well as ERK1/2 activation, with no effect on EGF-mediated ERK1/2 activation. Similar to G15, G36 inhibits estrogen- and G-1-stimulated proliferation of uterine epithelial cells in vivo. The identification of G36 as a GPER antagonist with improved ER counterselectivity represents a significant step towards the development of new highly selective therapeutics for cancer and other diseases.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures







References
-
- Edwards DP. Regulation of signal transduction pathways by estrogen and progesterone. Annu. Rev. Physiol. 2005;67:335–376. - PubMed
-
- Lange CA, Gioeli D, Hammes SR, Marker PC. Integration of rapid signaling events with steroid hormone receptor action in breast and prostate cancer. Annu. Rev. Physiol. 2007;69:171–199. - PubMed
-
- Prossnitz ER, Arterburn JB, Smith HO, Oprea TI, Sklar LA, Hathaway HJ. Estrogen signaling through the transmembrane G protein-coupled receptor GPR30. Annu. Rev. Physiol. 2008;70:165–190. - PubMed
-
- Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Ando S, Maggiolini M. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res. 2007;67(4):1859–1866. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
