

Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease
- PMID: 21785217
- PMCID: PMC3148745
- DOI: 10.1172/JCI57413
Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease
Abstract
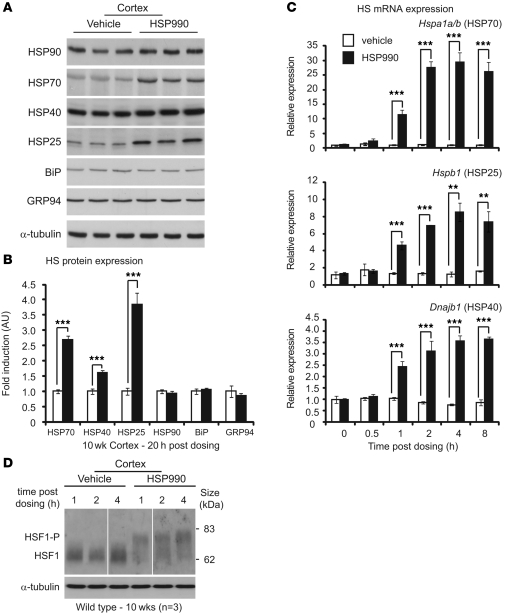
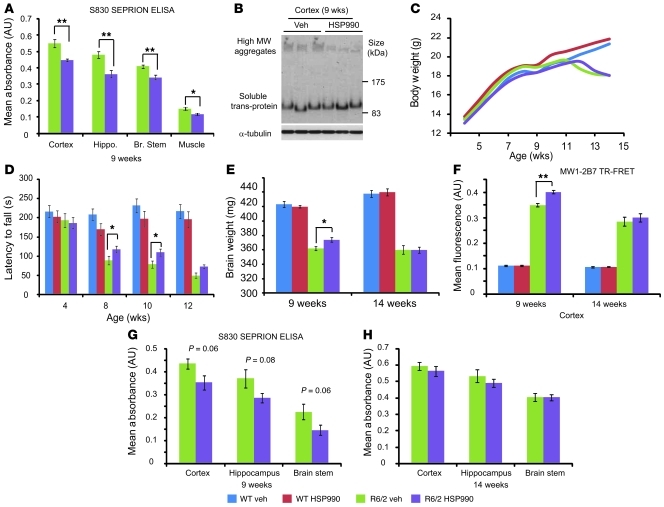
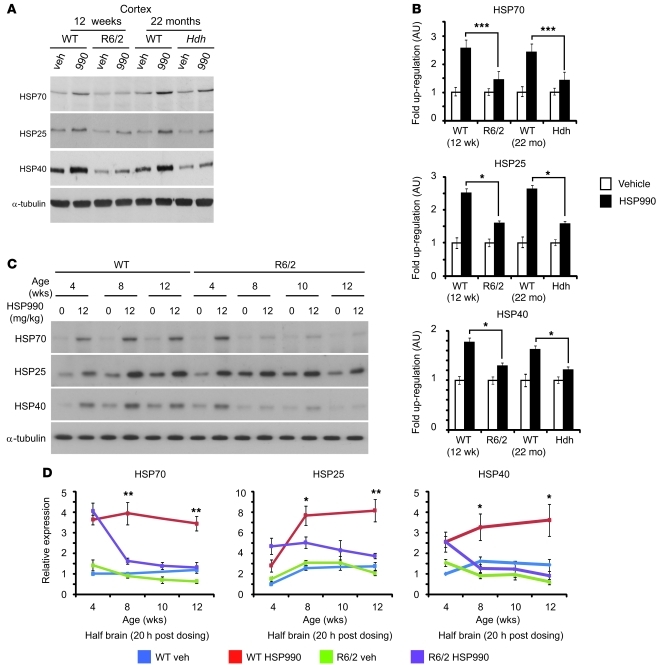
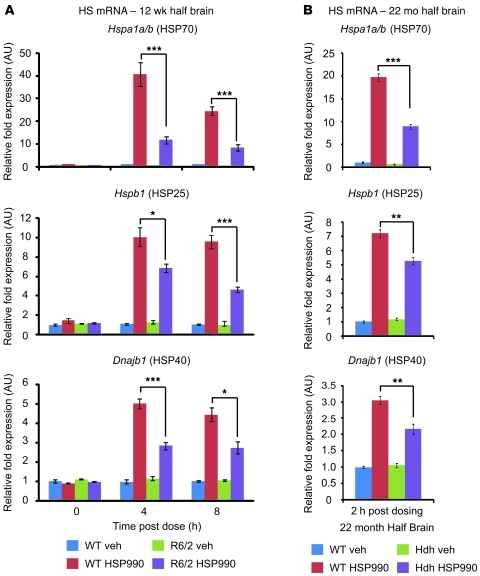
Huntington disease (HD) is a devastating neurodegenerative disorder for which there are no disease-modifying treatments. Previous studies have proposed that activation of the heat shock response (HSR) via the transcription factor heat shock factor 1 (HSF1) may be of therapeutic benefit. However, the effect of disease progression on the HSR and the therapeutic potential of this pathway are currently unknown. Here, we used a brain-penetrating HSP90 inhibitor and physiological, molecular, and behavioral readouts to demonstrate that pharmacological activation of HSF1 improves huntingtin aggregate load, motor performance, and other HD-related phenotypes in the R6/2 mouse model of HD. However, the beneficial effects of this treatment were transient and diminished with disease progression. Molecular analyses to understand the transient nature of these effects revealed altered chromatin architecture, reduced HSF1 binding, and impaired HSR accompanied disease progression in both the R6/2 transgenic and HdhQ150 knockin mouse models of HD. Taken together, our findings reveal that the HSR, a major inducible regulator of protein homeostasis and longevity, is disrupted in HD. Consequently, pharmacological induction of HSF1 as a therapeutic approach to HD is more complex than was previously anticipated.
Figures
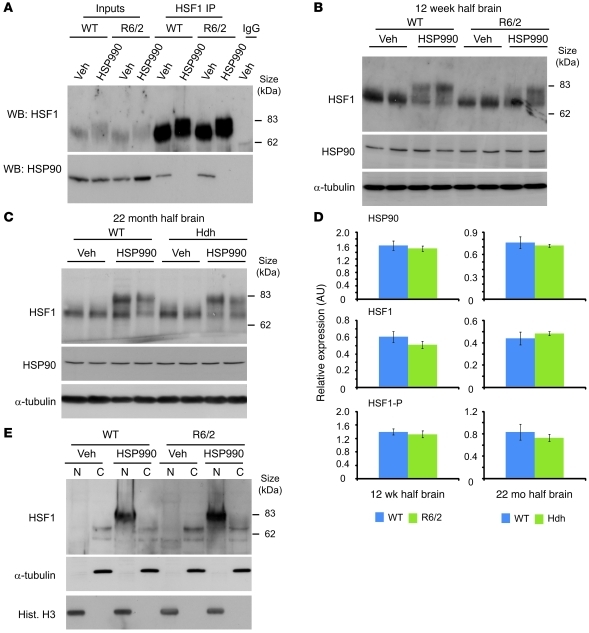

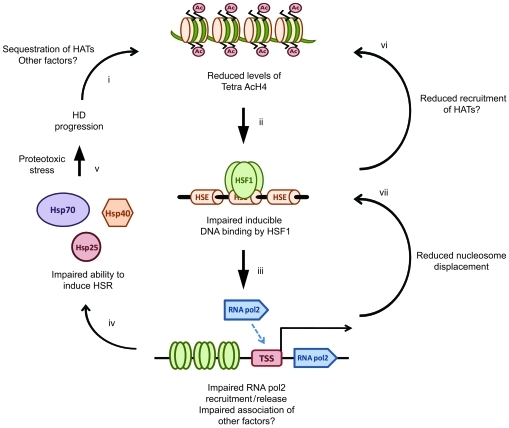
Comment in
-
Shock and awe: unleashing the heat shock response to treat Huntington disease.J Clin Invest. 2011 Aug;121(8):2972-5. doi: 10.1172/JCI59190. Epub 2011 Jul 25. J Clin Invest. 2011. PMID: 21785212 Free PMC article.
References
-
- Bates G, Harper P, Jones L, eds.Huntington’s Disease . 3rd ed. New York, New York, USA: Oxford University Press; 2002.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
