

Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy
- PMID: 21785219
- PMCID: PMC3148744
- DOI: 10.1172/JCI57291
Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy
Abstract
Spinal muscular atrophy (SMA) is a common neuromuscular disorder in humans. In fact, it is the most frequently inherited cause of infant mortality, being the result of mutations in the survival of motor neuron 1 (SMN1) gene that reduce levels of SMN protein. Restoring levels of SMN protein in individuals with SMA is perceived to be a viable therapeutic option, but the efficacy of such a strategy once symptoms are apparent has not been determined. We have generated mice harboring an inducible Smn rescue allele and used them in a model of SMA to investigate the effects of turning on SMN expression at different time points during the course of the disease. Restoring SMN protein even after disease onset was sufficient to reverse neuromuscular pathology and effect robust rescue of the SMA phenotype. Importantly, our findings also indicated that there was a therapeutic window of opportunity from P4 through P8 defined by the extent of neuromuscular synapse pathology and the ability of motor neurons to respond to SMN induction, following which restoration of the protein to the organism failed to produce therapeutic benefit. Nevertheless, our results suggest that even in severe SMA, timely reinstatement of the SMN protein may halt the progression of the disease and serve as an effective postsymptomatic treatment.
Figures
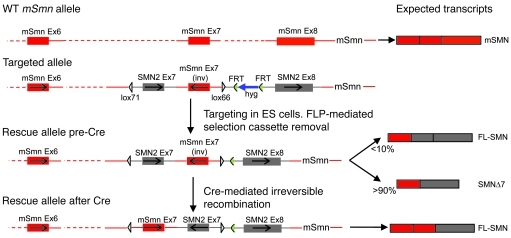
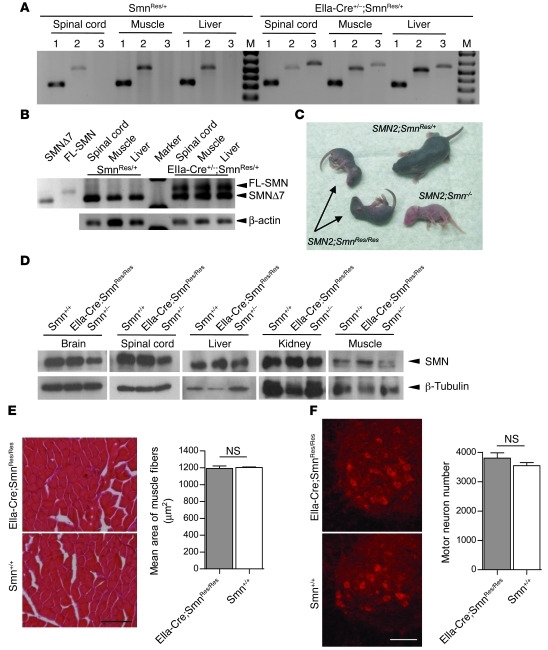
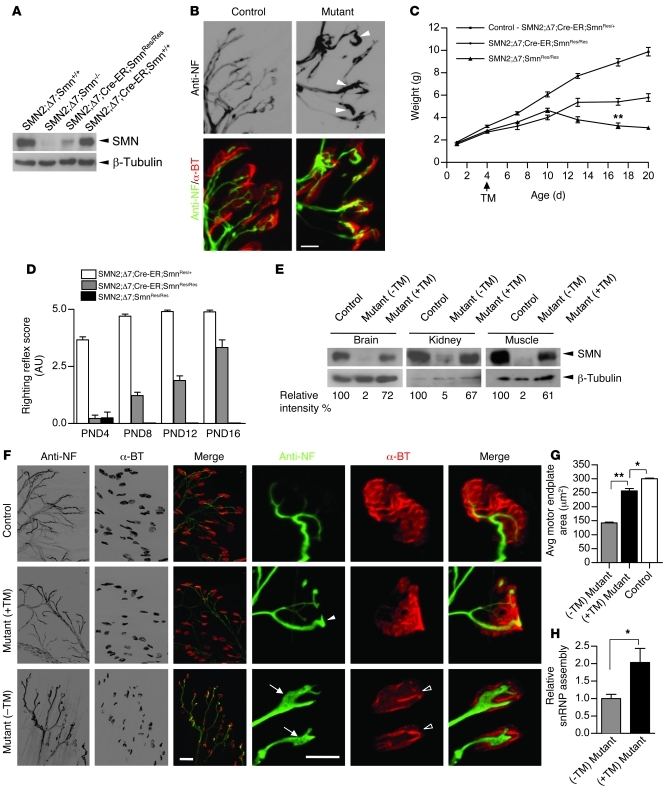
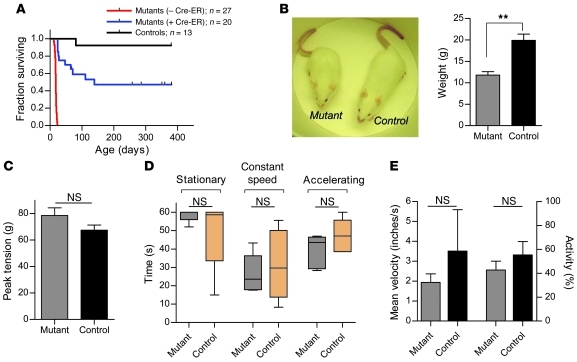
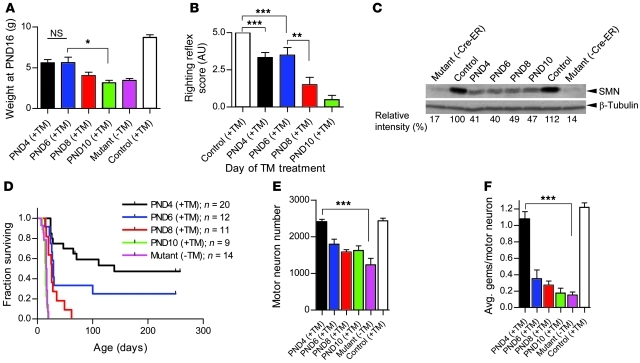
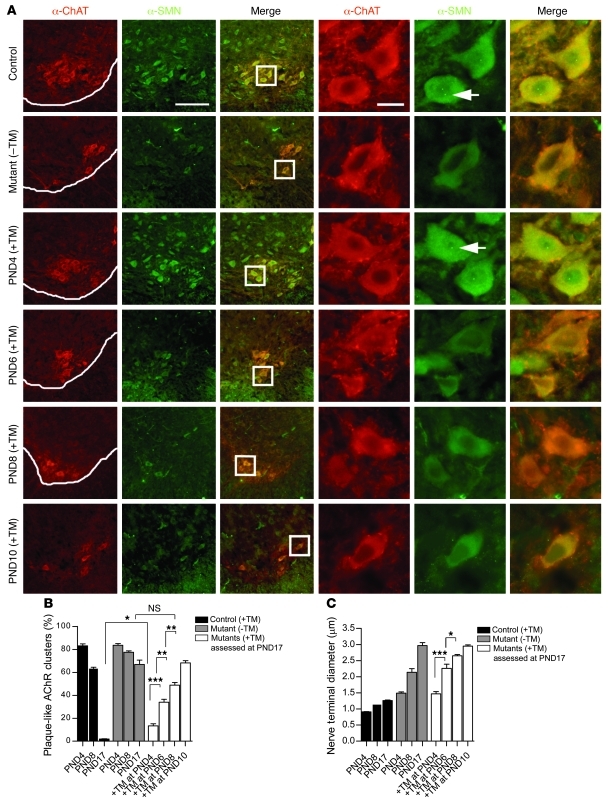
Comment in
-
Of SMN in mice and men: a therapeutic opportunity.J Clin Invest. 2011 Aug;121(8):2978-81. doi: 10.1172/JCI58752. Epub 2011 Jul 25. J Clin Invest. 2011. PMID: 21785213 Free PMC article.
References
-
- Lefebvre S, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16(3):265–269. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
