

Does amino acid sequence determine the properties of Aβ dimer?
- PMID: 21787025
- PMCID: PMC3155579
- DOI: 10.1063/1.3610427
Does amino acid sequence determine the properties of Aβ dimer?
Abstract
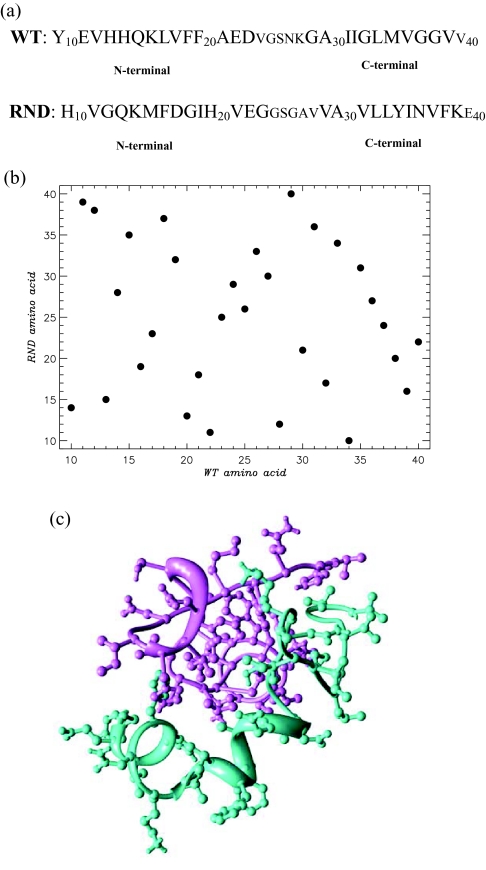
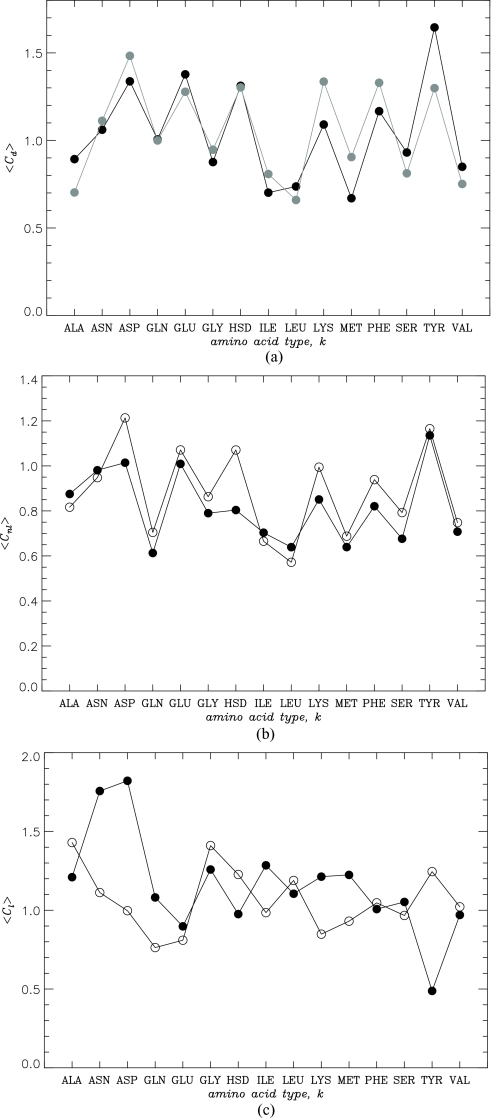
The effect of random reshuffling of amino acids on the properties of dimers formed by Aβ peptides is studied using replica exchange molecular dynamics and united atom implicit solvent model. We show that thermodynamics of dimer assembly and the dimer globule-like state are not affected by sequence permutation. Furthermore, sequence reshuffling does not change the distributions of non-local interactions and, to a large extent, amino acids in the dimer volume. To rationalize these results, we demonstrate that Gaussian statistics applies surprisingly well to the end-to-end distances of the peptides in the dimer implying that non-bonded interactions between distant along the chain amino acids are effectively screened. This observation suggests that peptides in the dimer behave as ideal chains in polymer melt, in which amino acids lose their "identity" and therefore the memory of sequence position. As a result large-scale properties of the dimer become universal or sequence independent. Comparison of our simulations with the prior theoretical studies and their implications for experiments are discussed.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials