

Analysis of human syndromes with disordered chromatin reveals the impact of heterochromatin on the efficacy of ATM-dependent G2/M checkpoint arrest
- PMID: 21791604
- PMCID: PMC3187363
- DOI: 10.1128/MCB.05289-11
Analysis of human syndromes with disordered chromatin reveals the impact of heterochromatin on the efficacy of ATM-dependent G2/M checkpoint arrest
Abstract
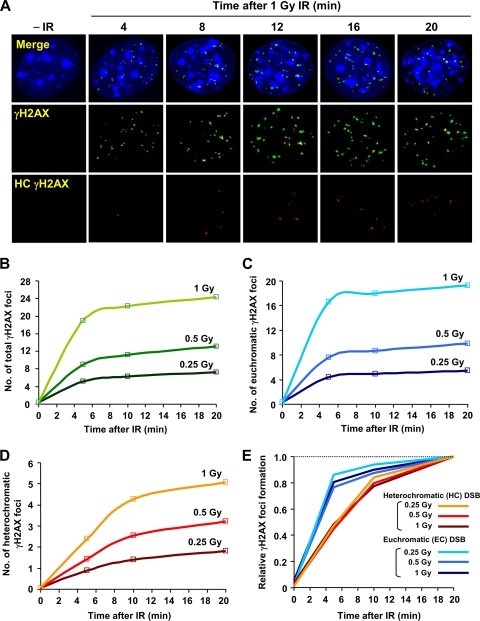
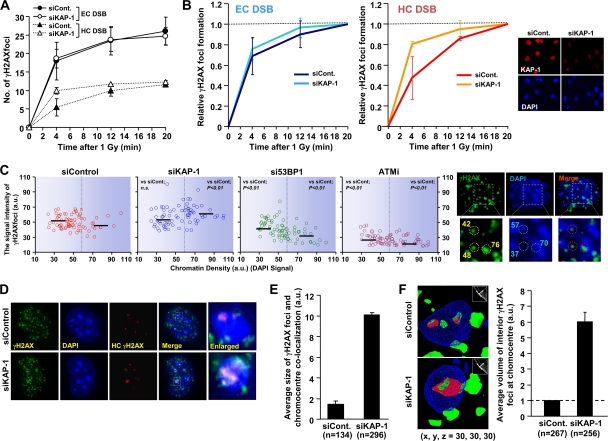
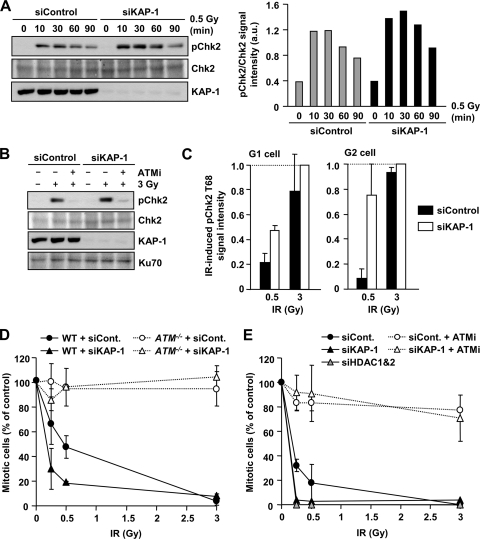
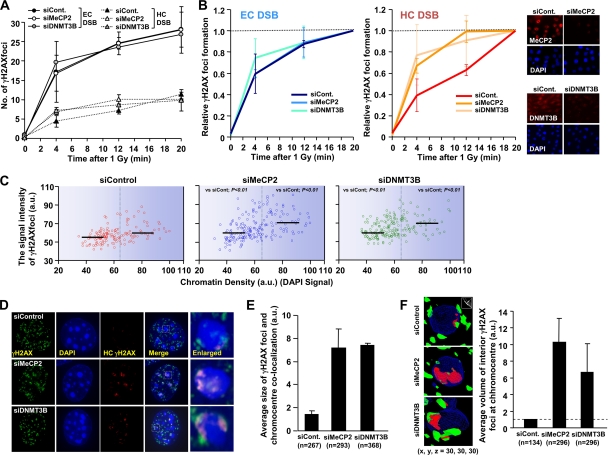
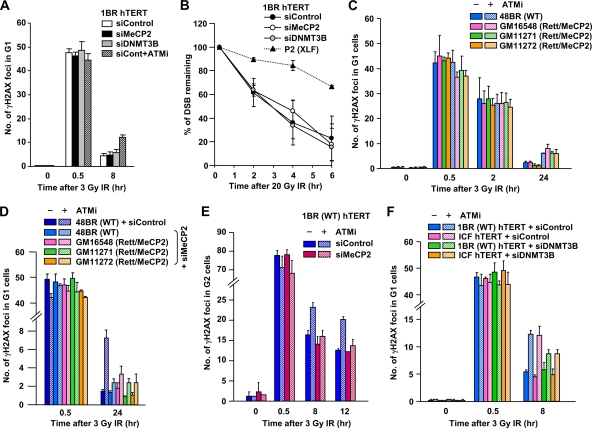
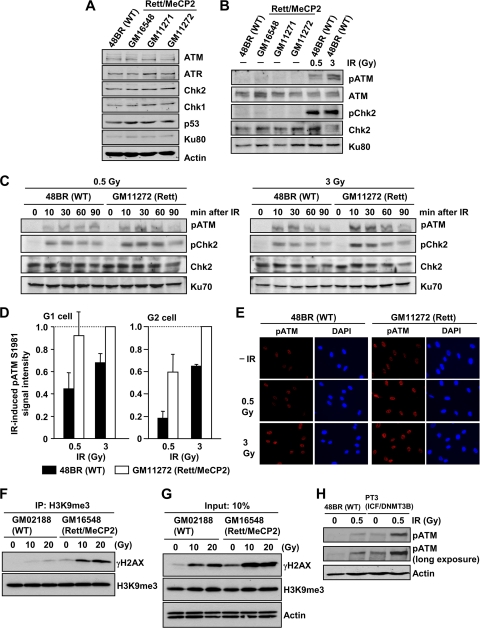
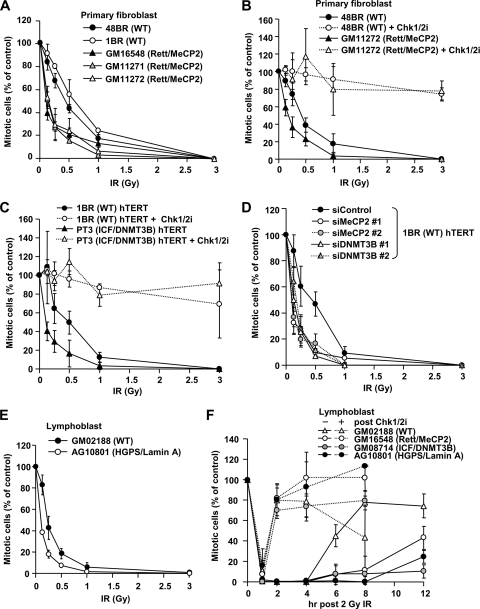
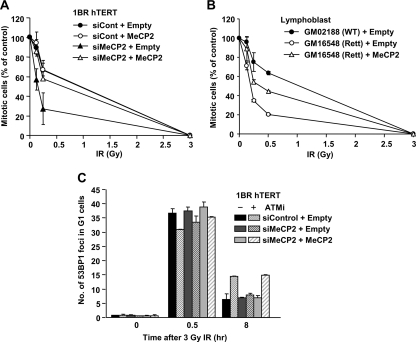
Heterochromatin (HC) poses a barrier to γH2AX focus expansion and DNA double-strand break (DSB) repair, the latter being relieved by ATM-dependent KAP-1 phosphorylation. Using high-resolution imaging, we show here that the HC superstructure markedly restricts ATM signaling to cell cycle checkpoint proteins. The impact of HC is greater than anticipated from the percentage of HC-DNA and, in distinction to DSB repair, ATM only partly overcomes the constraints posed by HC. Importantly, we examine ATM signaling in human syndromes with disordered HC. After depletion of MeCP2 and DNMT3B, proteins defective in the Rett and immunodeficiency with centromere instability and facial anomalies (ICF) syndromes, respectively, we demonstrate enhanced γH2AX signal expansion at HC-chromocenters in mouse NIH 3T3 cells, which have visible HC-chromocenters. Previous studies have shown that the G(2)/M checkpoint is inefficient requiring multiple DSBs to initiate arrest. MeCP2 and DNMT3B depletion leads to hypersensitive radiation-induced G(2)/M checkpoint arrest despite normal DSB repair. Cell lines from Rett, ICF, and Hutchinson-Guildford progeria syndrome patients similarly showed hyperactivated ATM signaling and hypersensitive and prolonged G(2)/M checkpoint arrest. Collectively, these findings reveal that heterochromatin contributes to the previously described inefficient G(2)/M checkpoint arrest and demonstrate how the signaling response can be uncoupled from DSB repair.
Figures
References
-
- Adams J. M., Cory S. 1998. The Bcl-2 protein family: arbiters of cell survival. Science 281:1322–1326 - PubMed
-
- Amir R. E., et al. 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23:185–188 - PubMed
-
- Anstey A. V., et al. 1999. Characterization of photosensitivity in the Smith-Lemli-Opitz syndrome: a new congenital photosensitivity syndrome. Br. J. Dermatol. 141:406–414 - PubMed
-
- Barber J. B., et al. 2000. Relationship between in vitro chromosomal radiosensitivity of peripheral blood lymphocytes and the expression of normal tissue damage following radiotherapy for breast cancer. Radiother. Oncol. 55:179–186 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous