

Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport
- PMID: 21794954
- PMCID: PMC3272324
- DOI: 10.1016/j.neurobiolaging.2011.06.006
Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport
Abstract
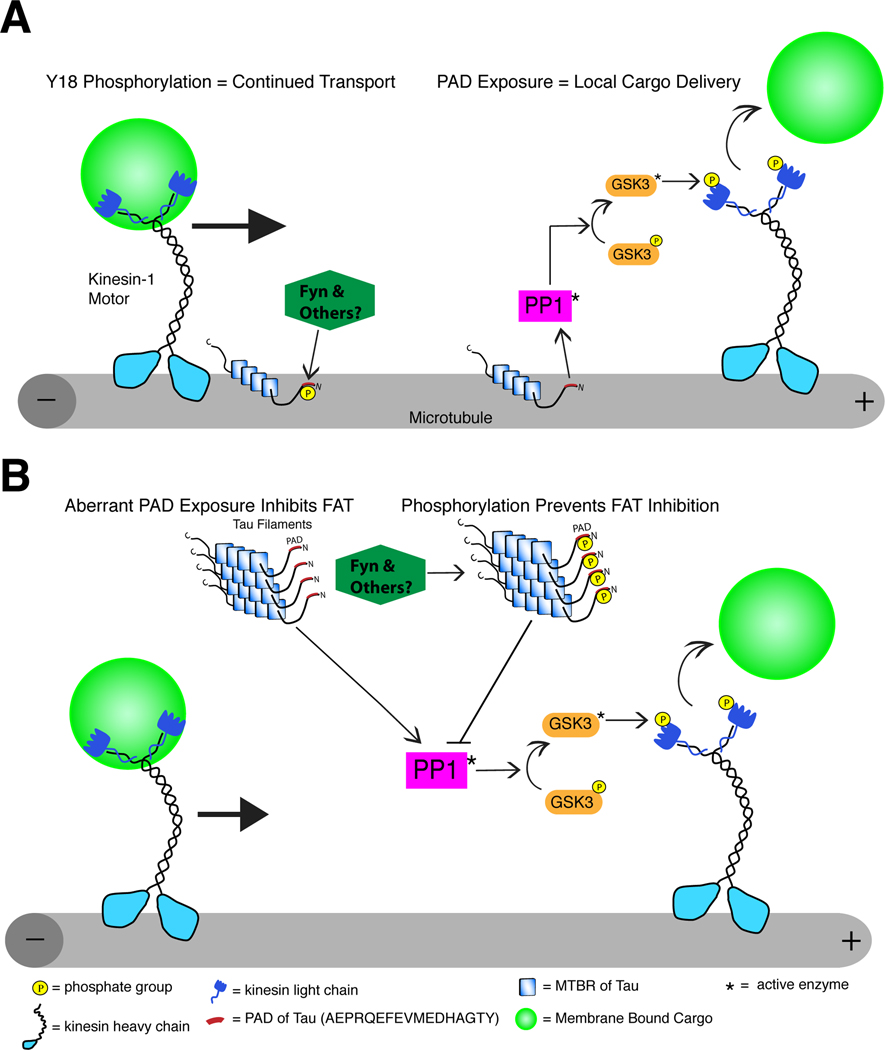
Alzheimer's disease (AD) and other tauopathies are characterized by fibrillar inclusions composed of the microtubule-associated protein, tau. Recently, we demonstrated that the N-terminus of tau (amino acids [aa] 2-18) in filamentous aggregates or N-terminal tau isoforms activate a signaling cascade involving protein phosphatase 1 and glycogen synthase kinase 3 that results in inhibition of anterograde fast axonal transport (FAT). We have termed the functional motif comprised of aa 2-18 in tau the phosphatase-activating domain (PAD). Here, we show that phosphorylation of tau at tyrosine 18, which is a fyn phosphorylation site within PAD, prevents inhibition of anterograde FAT induced by both filamentous tau and 6D tau. Moreover, Fyn-mediated phosphorylation of tyrosine 18 is reduced in disease-associated forms of tau (e.g., tau filaments). A novel PAD-specific monoclonal antibody revealed that exposure of PAD in tau occurs before and more frequently than tyrosine 18 phosphorylation in the evolution of tangle formation in AD. These results indicate that N-terminal phosphorylation may constitute a regulatory mechanism that controls tau-mediated inhibition of anterograde FAT in AD.
Copyright © 2012 Elsevier Inc. All rights reserved.
Conflict of interest statement
NMK, GM, STB, LIB and NEL are co-inventors on a pending patent for targeting PAD as therapeutic intervention for Alzheimer’s disease and other tauopathies. The authors have no other actual or potential conflicts of interest to report.
Figures
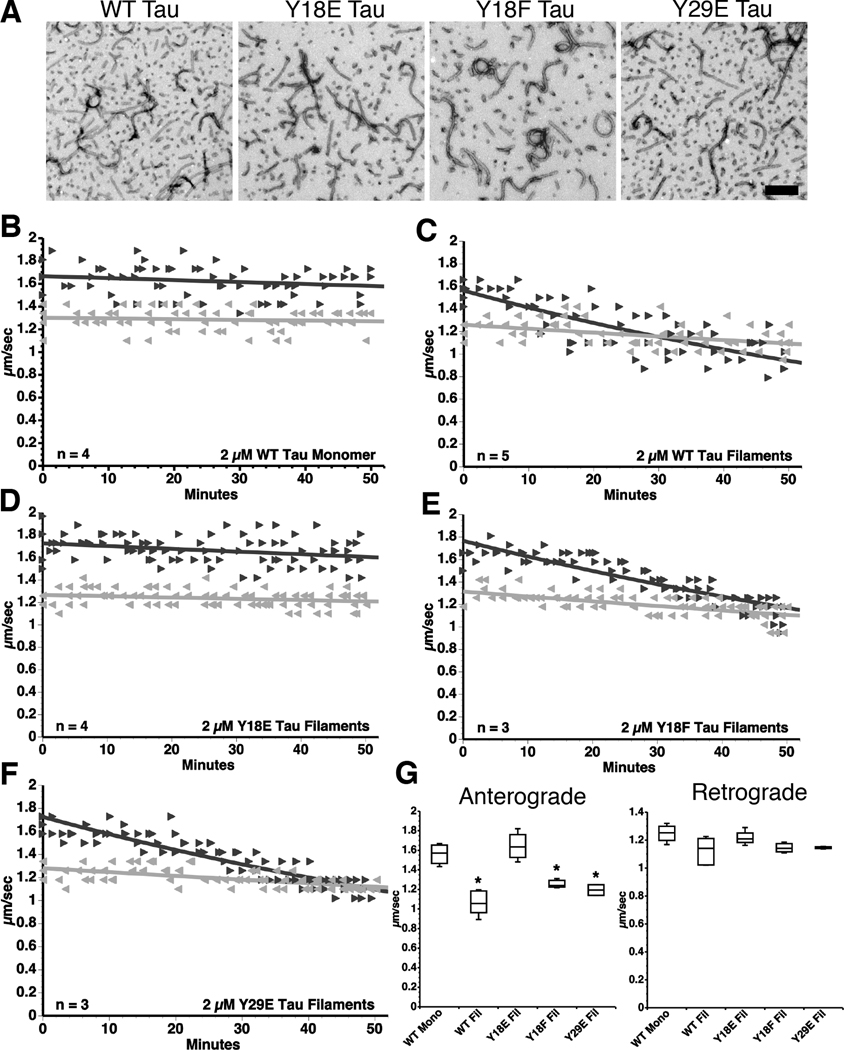
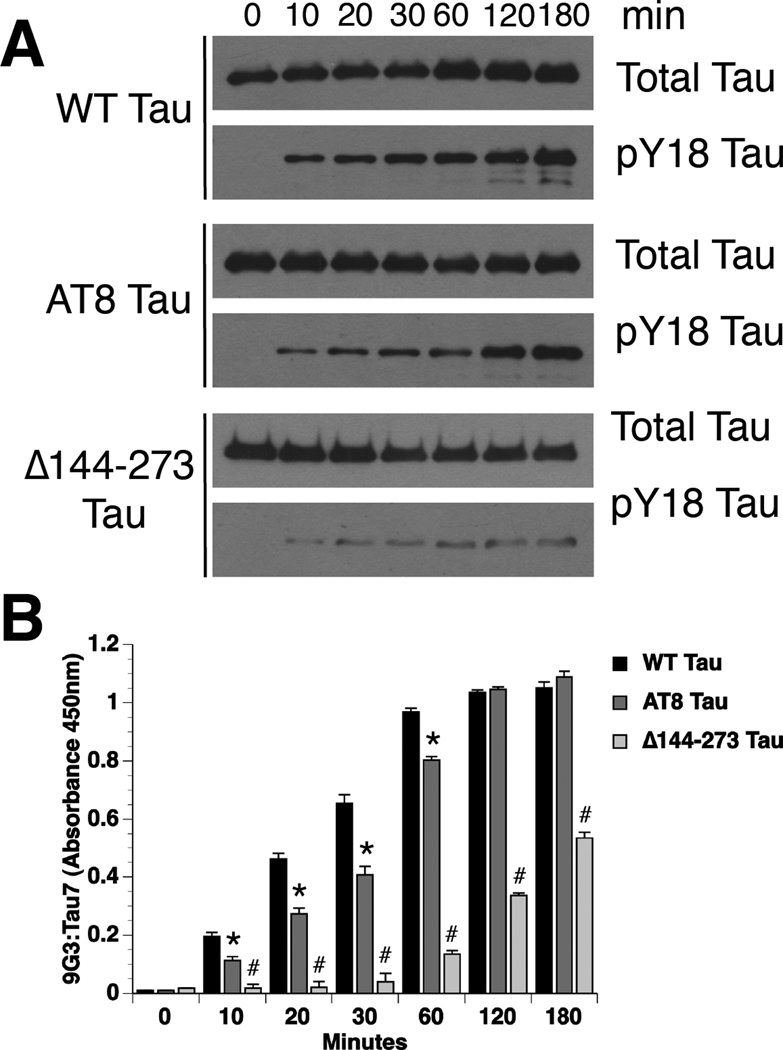
 ) FAT rates. (C) In contrast, perfusion of WT tau filaments selectively inhibited anterograde, but not retrograde FAT. (D) Filaments composed of Y18E tau did not affect FAT. (E) Y18F tau filaments (a non-phosphomimicking control mutation) selectively inhibited anterograde FAT (see Supplementary Fig. S1B). (F) Y29E tau filaments specifically decreased anterograde FAT. (G) Quantification of anterograde and retrograde FAT rates in B–F indicates that WT, Y18F and Y29E filaments significantly reduce the rate of anterograde FAT relative to WT tau monomers in isolated axoplasms (one-way ANOVA; * p<0.01). Retrograde FAT was unaffected by all tau constructs tested.
) FAT rates. (C) In contrast, perfusion of WT tau filaments selectively inhibited anterograde, but not retrograde FAT. (D) Filaments composed of Y18E tau did not affect FAT. (E) Y18F tau filaments (a non-phosphomimicking control mutation) selectively inhibited anterograde FAT (see Supplementary Fig. S1B). (F) Y29E tau filaments specifically decreased anterograde FAT. (G) Quantification of anterograde and retrograde FAT rates in B–F indicates that WT, Y18F and Y29E filaments significantly reduce the rate of anterograde FAT relative to WT tau monomers in isolated axoplasms (one-way ANOVA; * p<0.01). Retrograde FAT was unaffected by all tau constructs tested.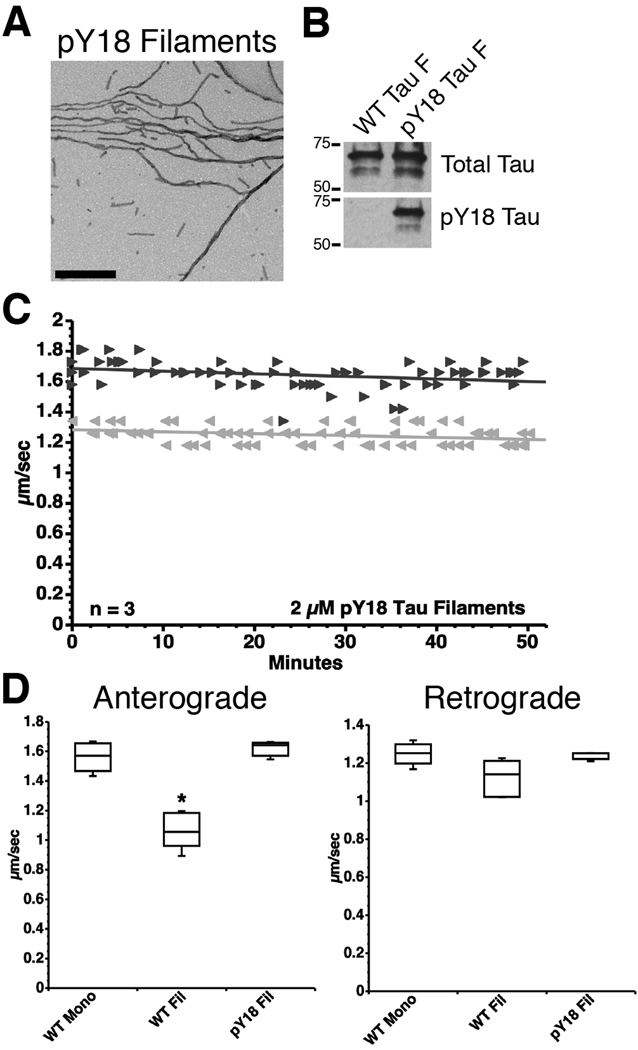 retrograde). (D) Box plots summarizing the statistical comparisons of data shown in panel C (one-way ANOVA; * p<0.01 compared to WT tau monomer and pY18 tau filaments).
retrograde). (D) Box plots summarizing the statistical comparisons of data shown in panel C (one-way ANOVA; * p<0.01 compared to WT tau monomer and pY18 tau filaments).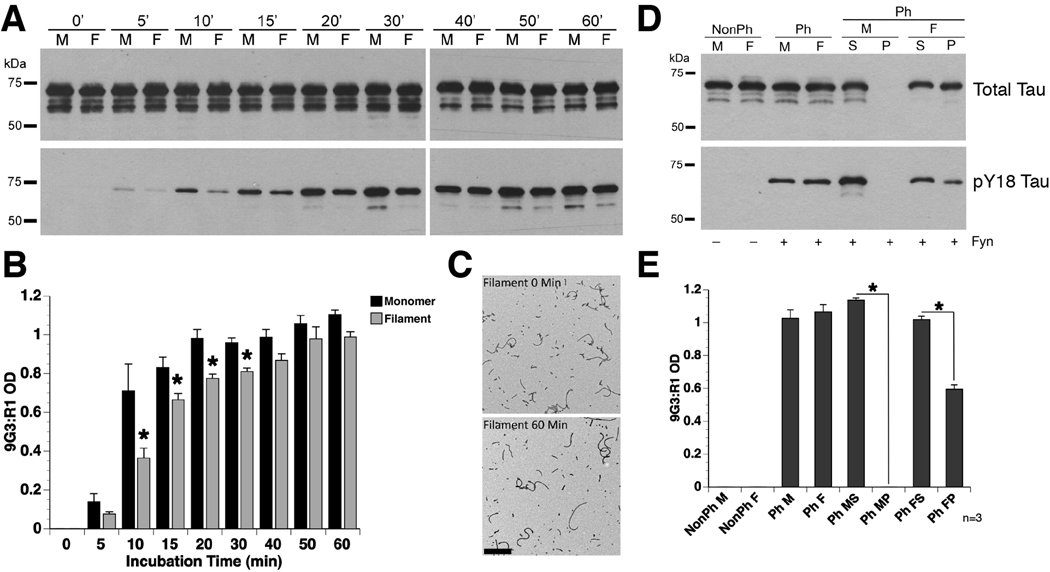
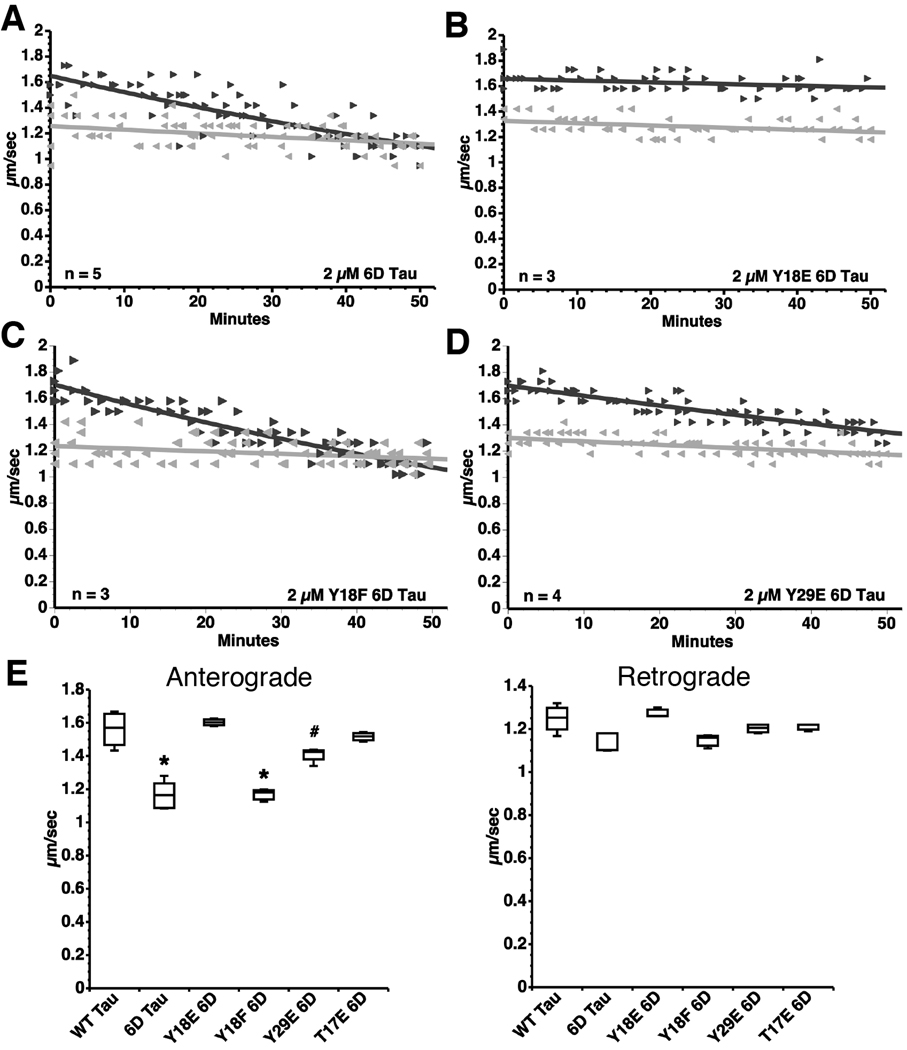 ). (B) Pseudophosphorylation at Y18 in 6D tau (Y18E) prevents this effect. (C) Perfusion of Y18F 6D tau inhibits anterograde FAT to an extent comparable to 6D tau. (D) Perfusion of Y29E 6D tau inhibits anterograde FAT to a lesser extent than 6D tau. (E) Box plots summarizing the data shown in panels A–D and the statistical comparisons (one-way ANOVA; * p<0.01 compared to WT tau, Y18E 6D, Y29E 6D and T17E 6D; # p<0.01 compared to Y18E 6D) (see also Supplementary Fig. S4).
). (B) Pseudophosphorylation at Y18 in 6D tau (Y18E) prevents this effect. (C) Perfusion of Y18F 6D tau inhibits anterograde FAT to an extent comparable to 6D tau. (D) Perfusion of Y29E 6D tau inhibits anterograde FAT to a lesser extent than 6D tau. (E) Box plots summarizing the data shown in panels A–D and the statistical comparisons (one-way ANOVA; * p<0.01 compared to WT tau, Y18E 6D, Y29E 6D and T17E 6D; # p<0.01 compared to Y18E 6D) (see also Supplementary Fig. S4).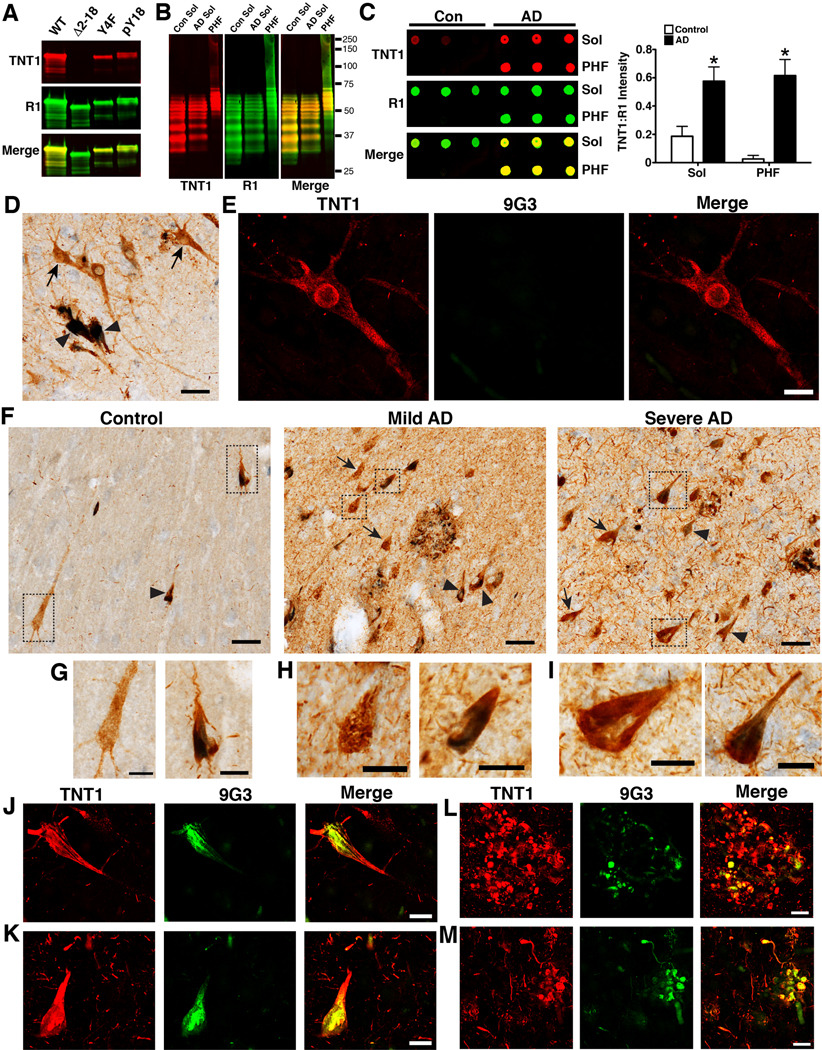
Similar articles
-
Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases.J Neurosci. 2011 Jul 6;31(27):9858-68. doi: 10.1523/JNEUROSCI.0560-11.2011. J Neurosci. 2011. PMID: 21734277 Free PMC article.
-
Pseudophosphorylation of tau at S422 enhances SDS-stable dimer formation and impairs both anterograde and retrograde fast axonal transport.Exp Neurol. 2016 Sep;283(Pt A):318-29. doi: 10.1016/j.expneurol.2016.06.030. Epub 2016 Jun 30. Exp Neurol. 2016. PMID: 27373205 Free PMC article.
-
Pathological conformations involving the amino terminus of tau occur early in Alzheimer's disease and are differentially detected by monoclonal antibodies.Neurobiol Dis. 2016 Oct;94:18-31. doi: 10.1016/j.nbd.2016.05.016. Epub 2016 May 31. Neurobiol Dis. 2016. PMID: 27260838 Free PMC article.
-
The microtubule-associated protein tau is also phosphorylated on tyrosine.J Alzheimers Dis. 2009;18(1):1-9. doi: 10.3233/JAD-2009-1116. J Alzheimers Dis. 2009. PMID: 19542604 Review.
-
Tau oligomers and tau toxicity in neurodegenerative disease.Biochem Soc Trans. 2012 Aug;40(4):667-71. doi: 10.1042/BST20120134. Biochem Soc Trans. 2012. PMID: 22817713 Free PMC article. Review.
Cited by
-
Components in SLPE Alleviate AD Model Nematodes by Up-Regulating Gene gst-5.Int J Mol Sci. 2024 Sep 23;25(18):10188. doi: 10.3390/ijms251810188. Int J Mol Sci. 2024. PMID: 39337674 Free PMC article.
-
Exposure of the Amino Terminus of Tau Is a Pathological Event in Multiple Tauopathies.Am J Pathol. 2017 Jun;187(6):1222-1229. doi: 10.1016/j.ajpath.2017.01.019. Epub 2017 Apr 14. Am J Pathol. 2017. PMID: 28413156 Free PMC article.
-
Fyn in Neurodevelopment and Ischemic Brain Injury.Dev Neurosci. 2015;37(4-5):311-20. doi: 10.1159/000369995. Epub 2015 Feb 17. Dev Neurosci. 2015. PMID: 25720756 Free PMC article. Review.
-
The pathogenic R5L mutation disrupts formation of Tau complexes on the microtubule by altering local N-terminal structure.Proc Natl Acad Sci U S A. 2022 Feb 15;119(7):e2114215119. doi: 10.1073/pnas.2114215119. Proc Natl Acad Sci U S A. 2022. PMID: 35135879 Free PMC article.
-
Tau phosphorylation and PAD exposure in regulation of axonal growth.Front Cell Dev Biol. 2023 Jan 18;10:1023418. doi: 10.3389/fcell.2022.1023418. eCollection 2022. Front Cell Dev Biol. 2023. PMID: 36742197 Free PMC article.
References
-
- Avila J, Hernandez F. GSK-3 inhibitors for Alzheimer's disease. Expert Rev Neurother. 2007;7:1527–1533. - PubMed
-
- Berry RW, Sweet AP, Clark FA, Lagalwar S, Lapin BR, Wang T, Topgi S, Guillozet-Bongaarts AL, Cochran EJ, Bigio EH, Binder LI. Tau epitope display in progressive supranuclear palsy and corticobasal degeneration. J Neurocytol. 2004;33:287–295. - PubMed
-
- Bhaskar K, Yen SH, Lee G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem. 2005;280:35119–35125. - PubMed
-
- Biernat J, Mandelkow EM, Schroter C, Lichtenberg-Kraag B, Steiner B, Berling B, Meyer H, Mercken M, Vandermeeren A, Goedert M. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992;11:1593–1597. - PMC - PubMed
-
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH., Jr Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
