

Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway
- PMID: 21795534
- PMCID: PMC6623085
- DOI: 10.1523/JNEUROSCI.1642-11.2011
Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway
Abstract
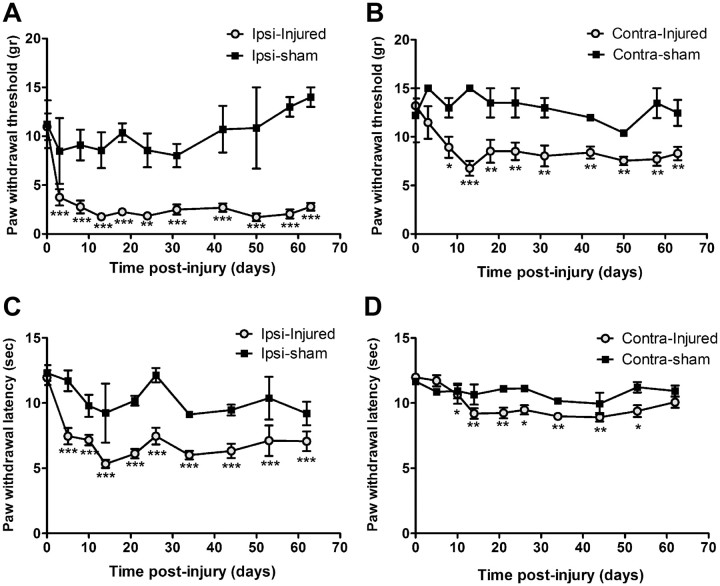
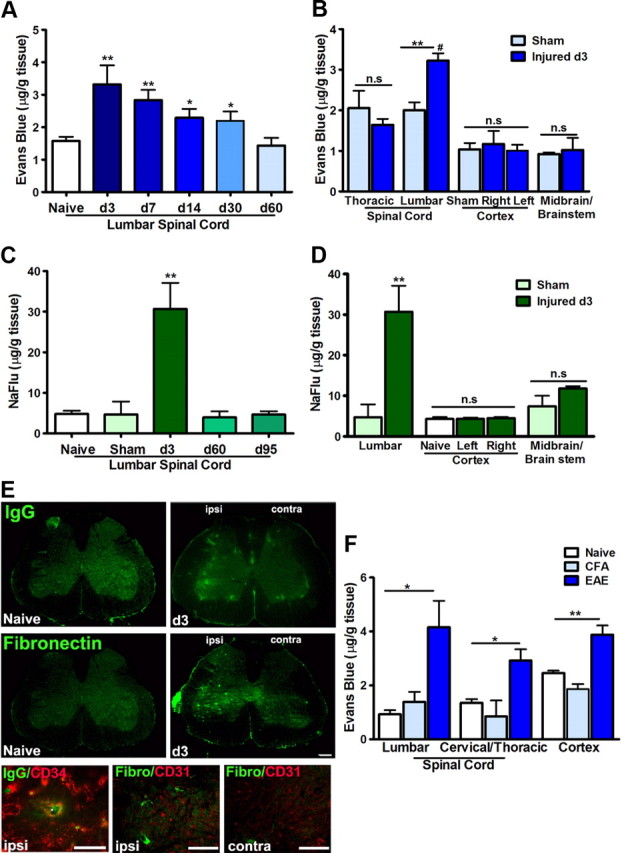
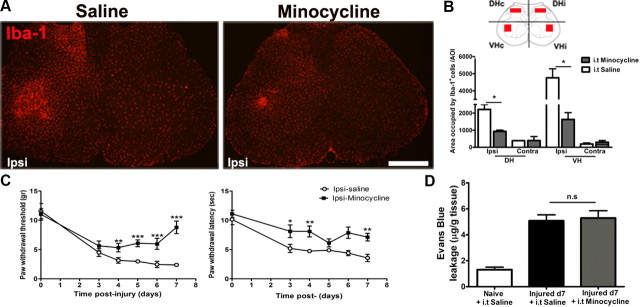
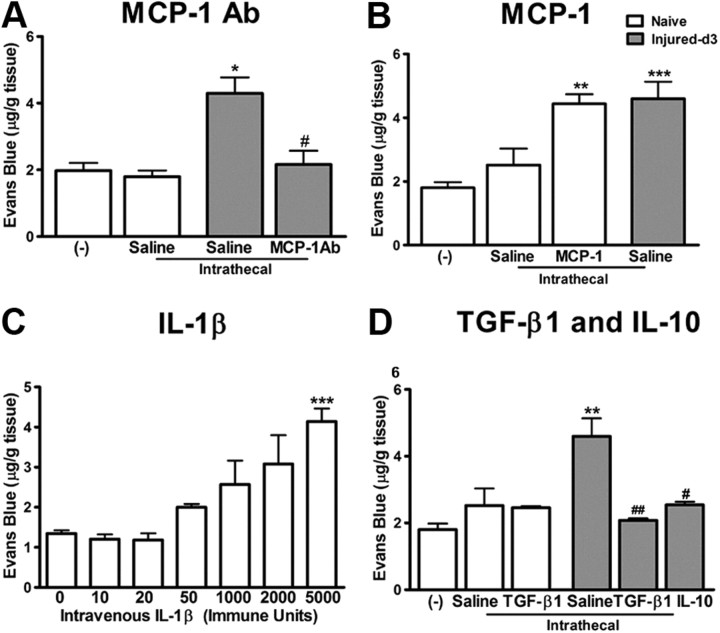
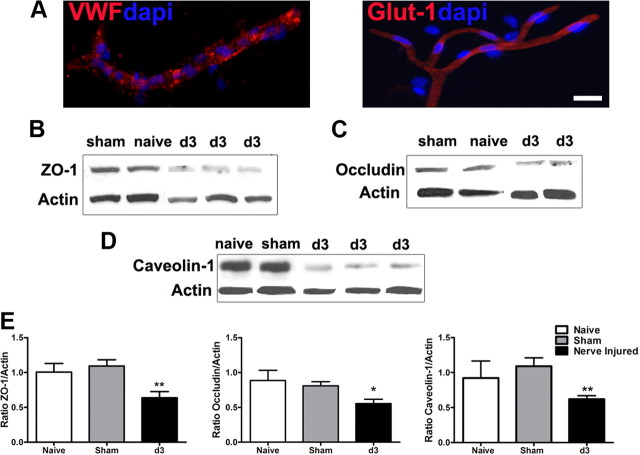
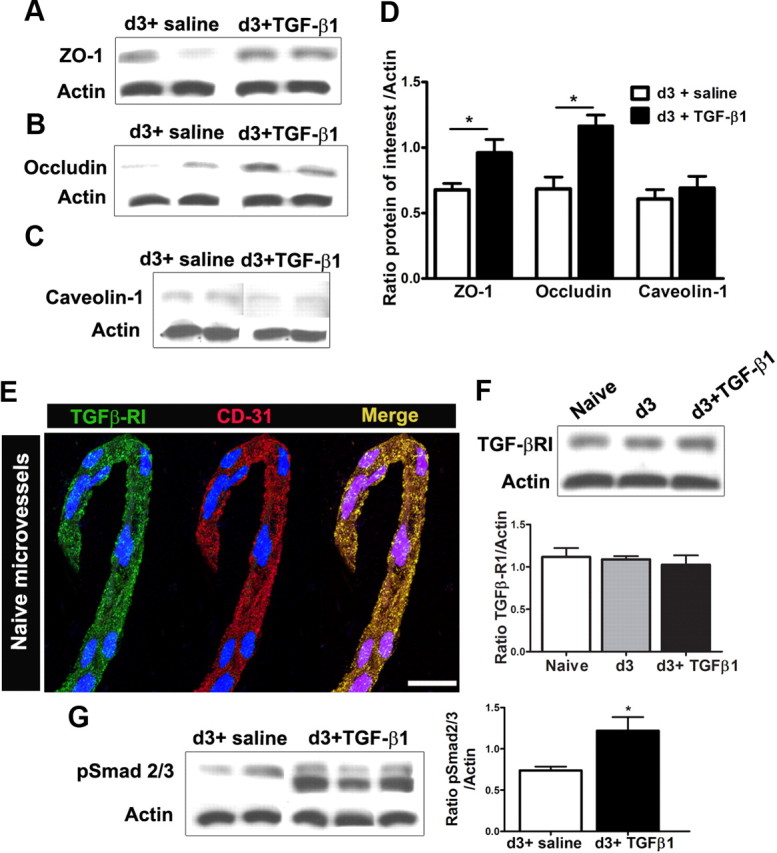
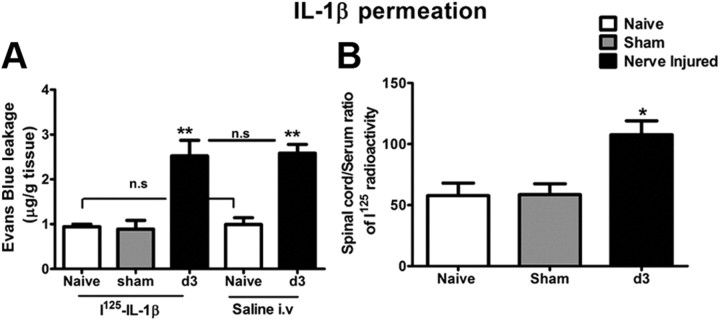
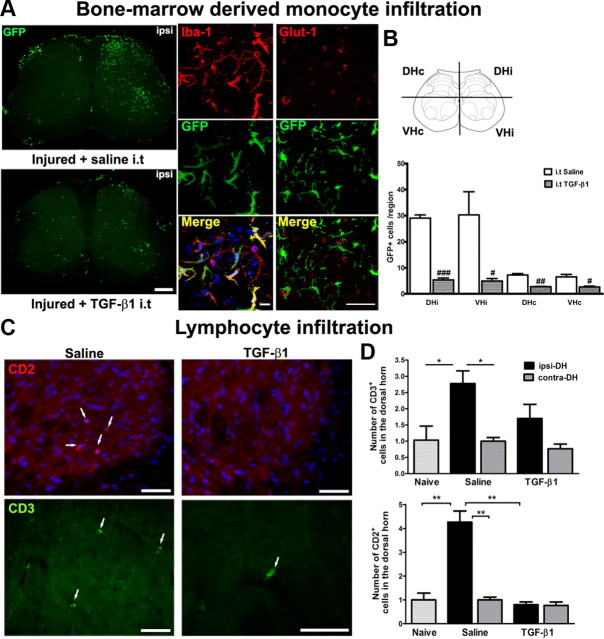
Peripheral nerve lesion triggers alterations in the spinal microenvironment that contribute to the pathogenesis of neuropathic pain. While neurons and glia have been implicated in these functional changes, it remains largely underexplored whether the blood-spinal cord barrier (BSCB) is also involved. The BSCB is an important component in the CNS homeostasis, and compromised BSCB has been associated with different pathologies affecting the spinal cord. Here, we demonstrated that a remote injury on the peripheral nerve in rats triggered a leakage of the BSCB, which was independent of spinal microglial activation. The increase of BSCB permeability to different size tracers, such as Evans Blue and sodium fluorescein, was restricted to the lumbar spinal cord and prominent for at least 4 weeks after injury. The spinal inflammatory reaction triggered by nerve injury was a key player in modulating BSCB permeability. We identified MCP-1 as an endogenous trigger for the BSCB leakage. BSCB permeability can also be impaired by circulating IL-1β. In contrast, antiinflammatory cytokines TGF-β1 and IL-10 were able to shut down the openings of the BSCB following nerve injury. Peripheral nerve injury caused a decrease in tight junction and caveolae-associated proteins. Interestingly, ZO-1 and occludin, but not caveolin-1, were rescued by TGF-β1. Furthermore, our data provide direct evidence that disrupted BSCB following nerve injury contributed to the influx of inflammatory mediators and the recruitment of spinal blood borne monocytes/macrophages, which played a major role in the development of neuropathic pain. These findings highlight the importance of inflammation in BSCB integrity and in spinal cord homeostasis.
Figures
References
-
- Banks WA, Erickson MA. The blood-brain barrier and immune function and dysfunction. Neurobiol Dis. 2010;37:26–32. - PubMed
-
- Bennett J, Basivireddy J, Kollar A, Biron KE, Reickmann P, Jefferies WA, McQuaid S. Blood-brain barrier disruption and enhanced vascular permeability in the multiple sclerosis model EAE. J Neuroimmunol. 2010;229:180–191. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous