

Deregulation of the ubiquitin-proteasome system is the predominant molecular pathology in OPMD animal models and patients
- PMID: 21798095
- PMCID: PMC3156638
- DOI: 10.1186/2044-5040-1-15
Deregulation of the ubiquitin-proteasome system is the predominant molecular pathology in OPMD animal models and patients
Abstract
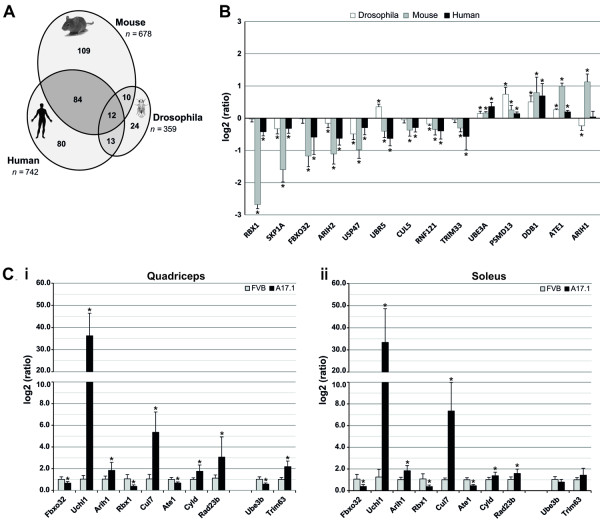
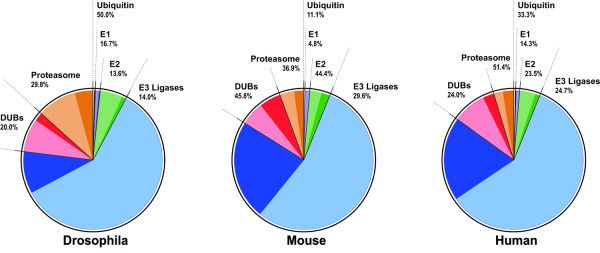
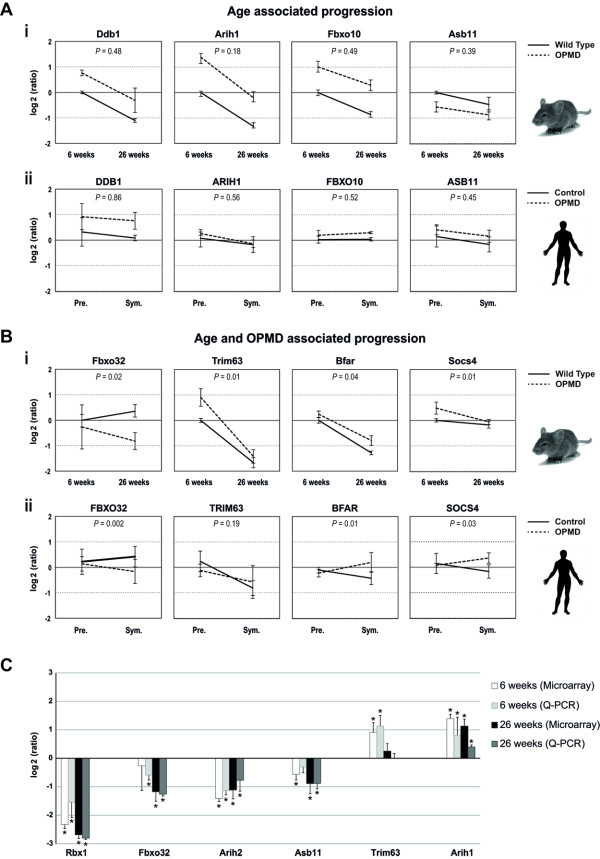
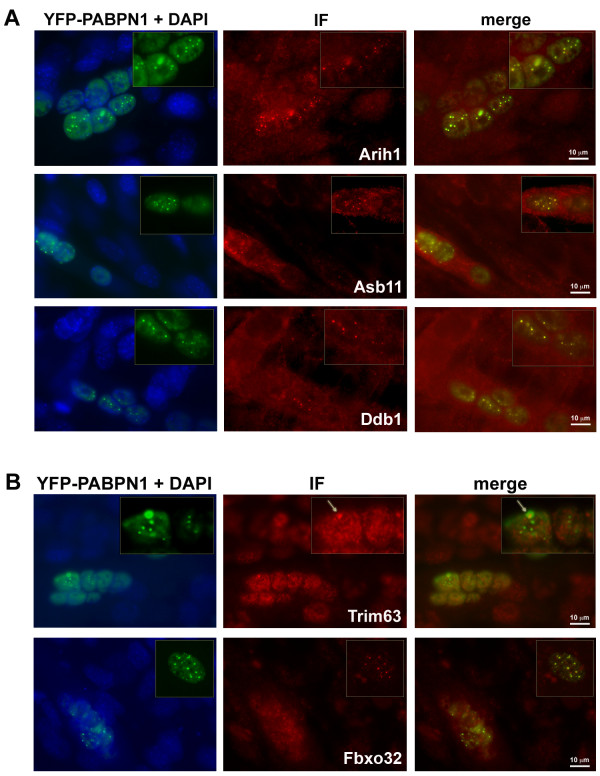
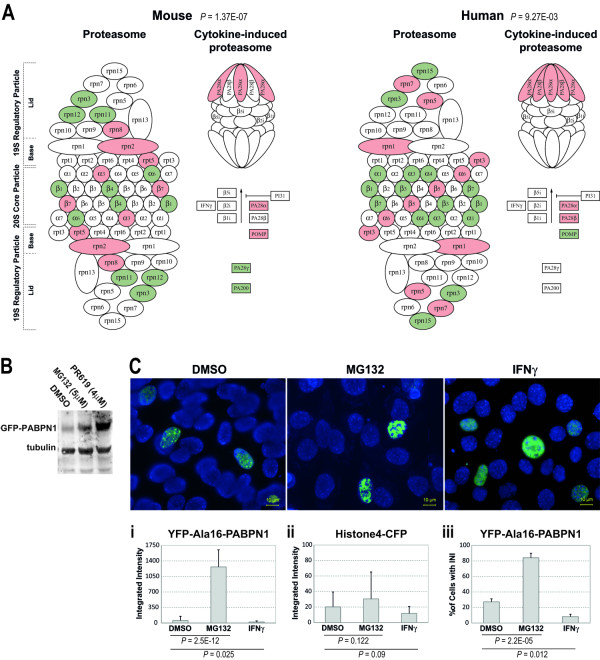
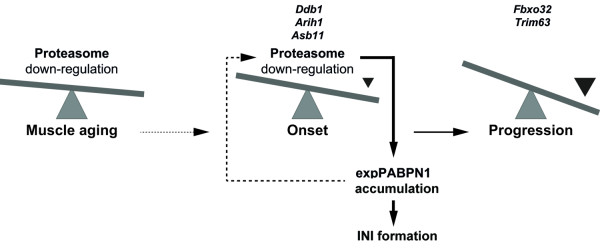
Oculopharyngeal muscular dystrophy (OPMD) is a late-onset progressive muscle disorder caused by a poly-alanine expansion mutation in the Poly(A) Binding Protein Nuclear 1 (PABPN1). The molecular mechanisms that regulate disease onset and progression are largely unknown. In order to identify molecular pathways that are consistently associated with OPMD, we performed an integrated high-throughput transcriptome study in affected muscles of OPMD animal models and patients. The ubiquitin-proteasome system (UPS) was found to be the most consistently and significantly OPMD-deregulated pathway across species. We could correlate the association of the UPS OPMD-deregulated genes with stages of disease progression. The expression trend of a subset of these genes is age-associated and therefore, marks the late onset of the disease, and a second group with expression trends relating to disease-progression. We demonstrate a correlation between expression trends and entrapment into PABPN1 insoluble aggregates of OPMD-deregulated E3 ligases. We also show that manipulations of proteasome and immunoproteasome activity specifically affect the accumulation and aggregation of mutant PABPN1. We suggest that the natural decrease in proteasome expression and its activity during muscle aging contributes to the onset of the disease.
Figures
References
-
- Fan X, Rouleau GA. Progress in understanding the pathogenesis of oculopharyngeal muscular dystrophy. Can J Neurol Sci. 2003;30:8–14. - PubMed
-
- Brais B, Xie YG, Sanson M, Morgan K, Weissenbach J, Korczyn AD, Blumen SC, Fardeau M, Tomé FM, Bouchard JP. et al. The oculopharyngeal muscular dystrophy locus maps to the region of the cardiac alpha and beta myosin heavy chain genes on chromosome 14q11.2-q13. Hum Mol Genet. 1995;4:429–434. doi: 10.1093/hmg/4.3.429. - DOI - PubMed
-
- Brais B, Bouchard JP, Xie YG, Rochefort DL, Chretien N, Tome FM, Lafrenière RG, Rommens JM, Uyama E, Nohira O, Blumen S, Korczyn AD, Heutink P, Mathieu J, Duranceau A, Codère F, Fardeau M, Rouleau GA. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat Genet. 1998;18:164–167. doi: 10.1038/ng0298-164. - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
