

From proteins to genes: immunoanalysis in the diagnosis of muscular dystrophies
- PMID: 21798100
- PMCID: PMC3156647
- DOI: 10.1186/2044-5040-1-24
From proteins to genes: immunoanalysis in the diagnosis of muscular dystrophies
Abstract
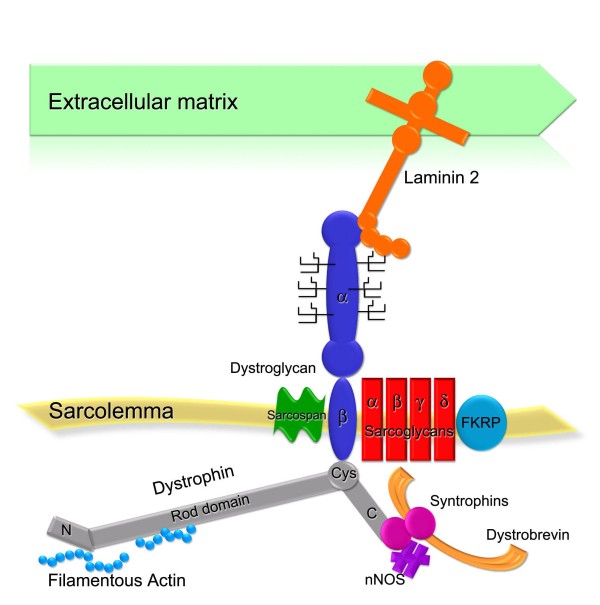
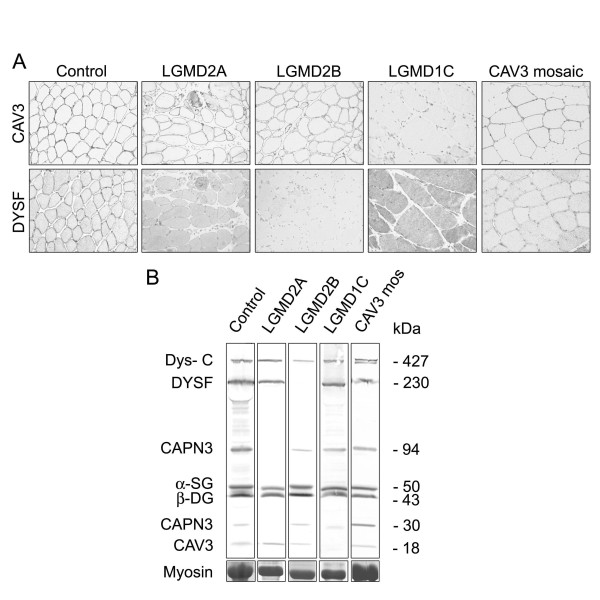
Muscular dystrophies are a large heterogeneous group of inherited diseases that cause progressive muscle weakness and permanent muscle damage. Very few muscular dystrophies show sufficient specific clinical features to allow a definite diagnosis. Because of the currently limited capacity to screen for numerous genes simultaneously, muscle biopsy is a time and cost-effective test for many of these disorders. Protein analysis interpreted in correlation with the clinical phenotype is a useful way of directing genetic testing in many types of muscular dystrophies. Immunohistochemistry and western blot are complementary techniques used to gather quantitative and qualitative information on the expression of proteins involved in this group of diseases. Immunoanalysis has a major diagnostic application mostly in recessive conditions where the absence of labelling for a particular protein is likely to indicate a defect in that gene. However, abnormalities in protein expression can vary from absence to very subtle reduction. It is good practice to test muscle biopsies with antibodies for several proteins simultaneously and to interpret the results in context. Indeed, there is a degree of direct or functional association between many of these proteins that is reflected by the presence of specific secondary abnormalities that are of value, especially when the diagnosis is not straightforward.
Figures


Similar articles
-
Strategy for mutation analysis in the autosomal recessive limb-girdle muscular dystrophies.Neuromuscul Disord. 2001 Jan;11(1):80-7. doi: 10.1016/s0960-8966(00)00154-1. Neuromuscul Disord. 2001. PMID: 11166169
-
Muscular dystrophies: an update on pathology and diagnosis.Acta Neuropathol. 2010 Sep;120(3):343-58. doi: 10.1007/s00401-010-0727-5. Epub 2010 Jul 23. Acta Neuropathol. 2010. PMID: 20652576 Review.
-
Protein Expression of Canine and Feline Muscular Dystrophies.Top Companion Anim Med. 2021 Mar;42:100500. doi: 10.1016/j.tcam.2020.100500. Epub 2020 Nov 26. Top Companion Anim Med. 2021. PMID: 33249241
-
Histochemical and immunohistological approach to comparative neuromuscular diseases.Folia Histochem Cytobiol. 2009;47(2):143-52. doi: 10.2478/v10042-009-0066-3. Folia Histochem Cytobiol. 2009. PMID: 19995699 Review.
-
[Muscular dystrophies detected by immunophenotyping and genotype analysis (mRNA and DNA)].Cesk Patol. 2001 Nov;37(4):137-45. Cesk Patol. 2001. PMID: 11813630 Czech.
Cited by
-
Exercise-induced alterations and loss of sarcomeric M-line organization in the diaphragm muscle of obscurin knockout mice.Am J Physiol Cell Physiol. 2017 Jan 1;312(1):C16-C28. doi: 10.1152/ajpcell.00098.2016. Epub 2016 Oct 26. Am J Physiol Cell Physiol. 2017. PMID: 27784675 Free PMC article.
-
Improved efficacy of a next-generation ERT in murine Pompe disease.JCI Insight. 2019 Mar 7;4(5):e125358. doi: 10.1172/jci.insight.125358. eCollection 2019 Mar 7. JCI Insight. 2019. PMID: 30843882 Free PMC article.
-
The Classification, Natural History and Treatment of the Limb Girdle Muscular Dystrophies.J Neuromuscul Dis. 2015 Jul 22;2(s2):S7-S19. doi: 10.3233/JND-150105. J Neuromuscul Dis. 2015. PMID: 27858764 Free PMC article.
-
Exome sequences versus sequential gene testing in the UK highly specialised Service for Limb Girdle Muscular Dystrophy.Orphanet J Rare Dis. 2017 Sep 6;12(1):151. doi: 10.1186/s13023-017-0699-9. Orphanet J Rare Dis. 2017. PMID: 28877744 Free PMC article.
-
Predominance of Dystrophinopathy Genotypes in Mexican Male Patients Presenting as Muscular Dystrophy with A Normal Multiplex Polymerase Chain Reaction DMD Gene Result: A Study Including Targeted Next-Generation Sequencing.Genes (Basel). 2019 Oct 29;10(11):856. doi: 10.3390/genes10110856. Genes (Basel). 2019. PMID: 31671740 Free PMC article.
References
-
- Gasper MC, Gilchrist JM. Creatine kinase: a review of its use in the diagnosis of muscle disease. Med Health R I. 2005;88:398. 400-394. - PubMed
-
- M Mercuri E, Bushby K, Ricci E, Birchall D, Pane M, Kinali M, Allsop J, Nigro V, Sáenz A, Nascimbeni A, Fulizio L, Angelini C, Muntoni F. Muscle MRI findings in patients with limb girdle muscular dystrophy with calpain 3 deficiency (LGMD2A) and early contractures. Neuromuscul Disord. 2005;15:164–171. doi: 10.1016/j.nmd.2004.10.008. - DOI - PubMed
-
- Mercuri E, Pichiecchio A, Allsop J, Messina S, Pane M, Muntoni F. Muscle MRI in inherited neuromuscular disorders: past, present, and future. J Magn Reson Imaging. 2007;25:433–440. - PubMed
LinkOut - more resources
Full Text Sources
