

Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates
- PMID: 21799664
- PMCID: PMC3136466
- DOI: 10.1371/journal.ppat.1002132
Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates
Abstract
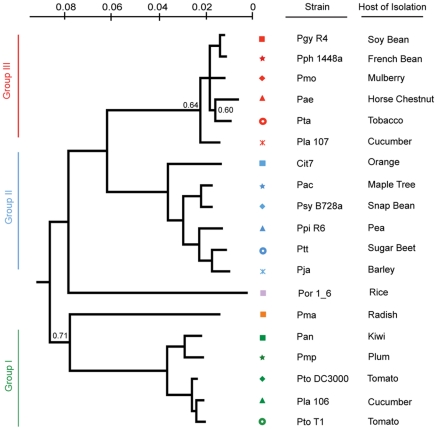
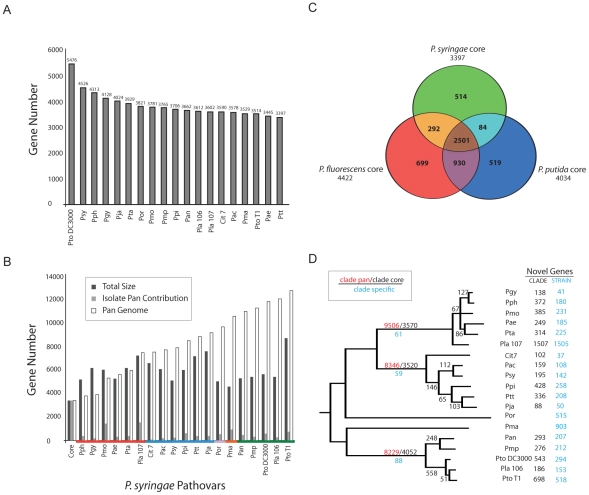
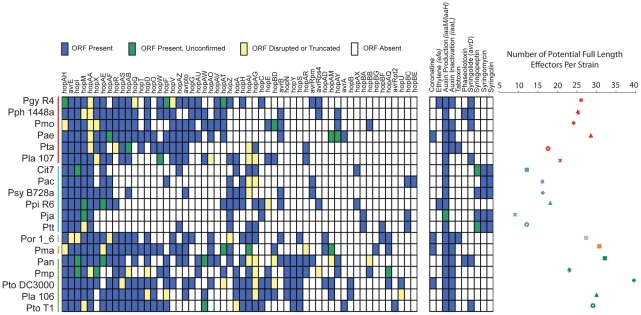
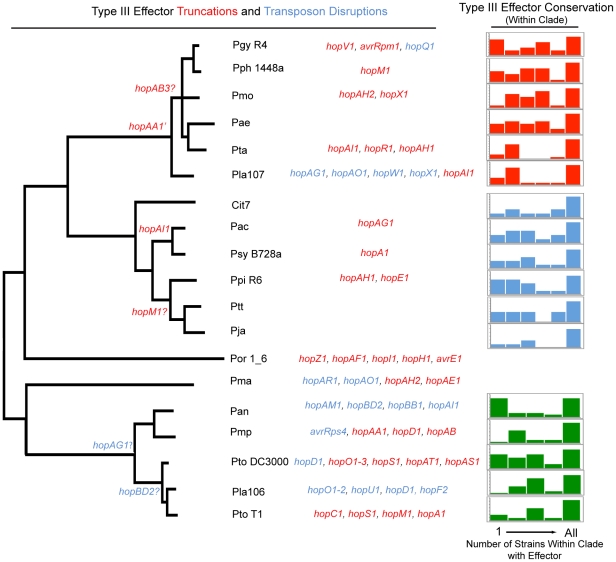
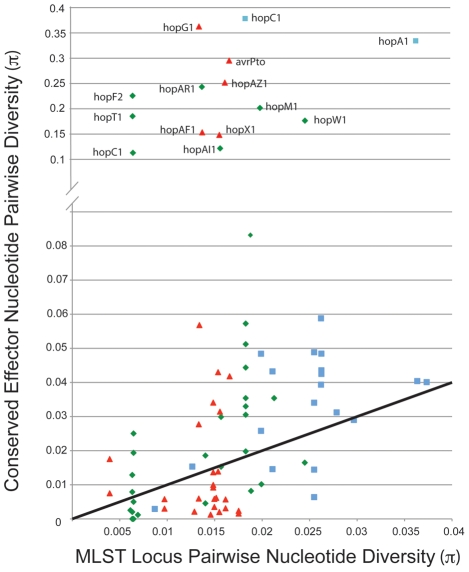
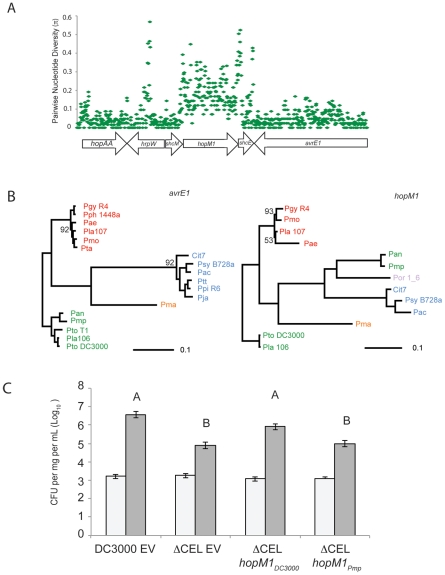
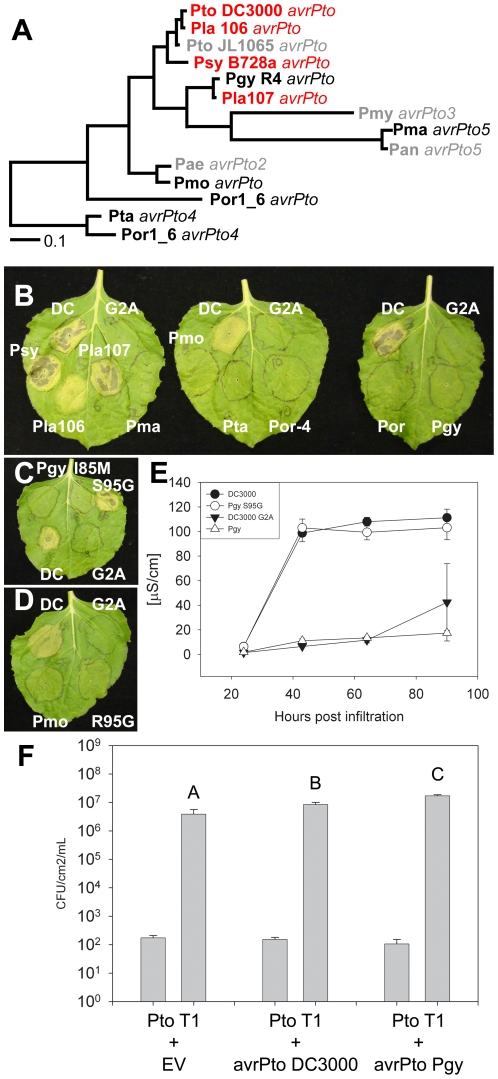
Closely related pathogens may differ dramatically in host range, but the molecular, genetic, and evolutionary basis for these differences remains unclear. In many Gram- negative bacteria, including the phytopathogen Pseudomonas syringae, type III effectors (TTEs) are essential for pathogenicity, instrumental in structuring host range, and exhibit wide diversity between strains. To capture the dynamic nature of virulence gene repertoires across P. syringae, we screened 11 diverse strains for novel TTE families and coupled this nearly saturating screen with the sequencing and assembly of 14 phylogenetically diverse isolates from a broad collection of diseased host plants. TTE repertoires vary dramatically in size and content across all P. syringae clades; surprisingly few TTEs are conserved and present in all strains. Those that are likely provide basal requirements for pathogenicity. We demonstrate that functional divergence within one conserved locus, hopM1, leads to dramatic differences in pathogenicity, and we demonstrate that phylogenetics-informed mutagenesis can be used to identify functionally critical residues of TTEs. The dynamism of the TTE repertoire is mirrored by diversity in pathways affecting the synthesis of secreted phytotoxins, highlighting the likely role of both types of virulence factors in determination of host range. We used these 14 draft genome sequences, plus five additional genome sequences previously reported, to identify the core genome for P. syringae and we compared this core to that of two closely related non-pathogenic pseudomonad species. These data revealed the recent acquisition of a 1 Mb megaplasmid by a sub-clade of cucumber pathogens. This megaplasmid encodes a type IV secretion system and a diverse set of unknown proteins, which dramatically increases both the genomic content of these strains and the pan-genome of the species.
© 2011 Baltrus et al.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Fatmi MB, Collmer A, Sante Iacobellis N, editors. Springer; 2008. Pseudomonas syringae Pathovars and Related Pathogens - Identification, Epidemiology, and Genomics.433
-
- Lin N-C, Martin GB. An avrPto/avrPtoB mutant of Pseudomonas syringae pv. tomato DC3000 does not elicit Pto-mediated resistance and is less virulent on tomato. MPMI. 2005;18:43–51. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
