

A genome-wide comparative evolutionary analysis of herpes simplex virus type 1 and varicella zoster virus
- PMID: 21799886
- PMCID: PMC3143153
- DOI: 10.1371/journal.pone.0022527
A genome-wide comparative evolutionary analysis of herpes simplex virus type 1 and varicella zoster virus
Abstract
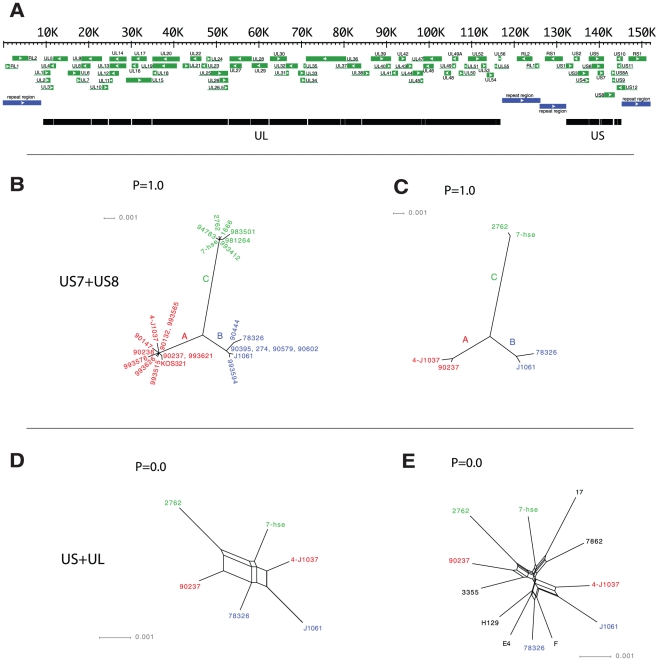
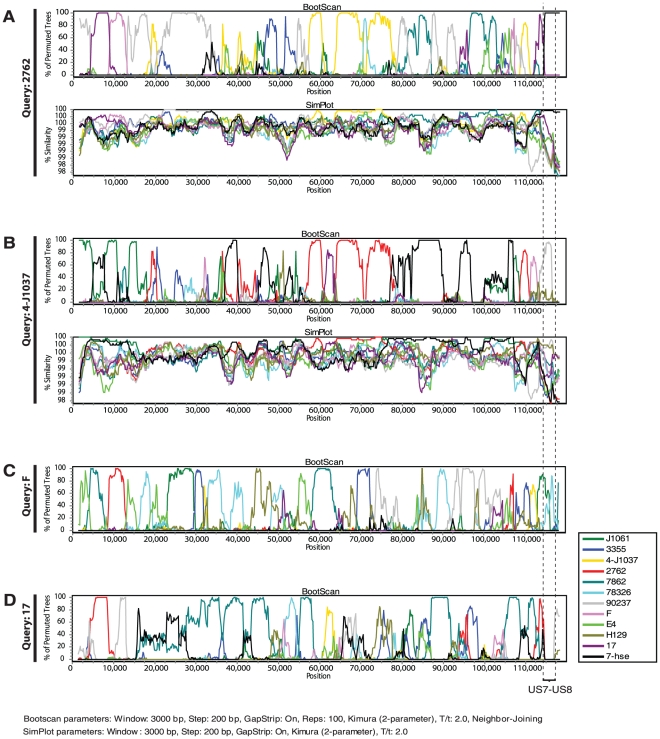
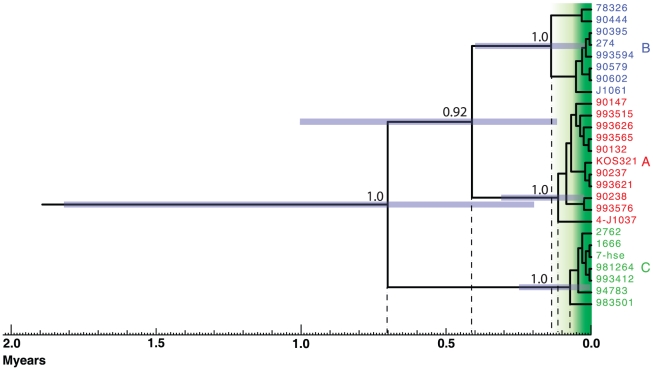
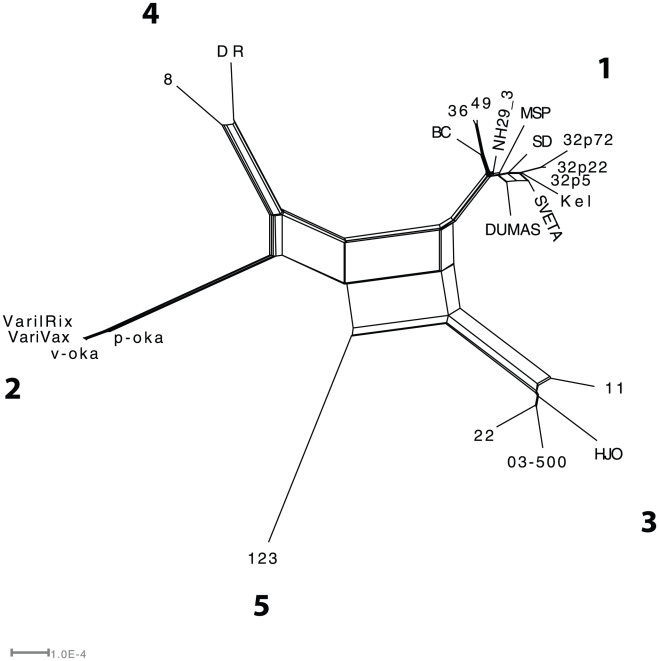
Herpes simplex virus type 1 (HSV-1) and varicella zoster virus (VZV) are closely related viruses causing lifelong infections. They are typically associated with mucocutaneous or skin lesions, but may also cause severe neurological or ophthalmic diseases, possibly due to viral- and/or host-genetic factors. Although these viruses are well characterized, genome-wide evolutionary studies have hitherto only been presented for VZV. Here, we present a genome-wide study on HSV-1. We also compared the evolutionary characteristics of HSV-1 with those for VZV. We demonstrate that, in contrast to VZV for which only a few ancient recombination events have been suggested, all HSV-1 genomes contain mosaic patterns of segments with different evolutionary origins. Thus, recombination seems to occur extremely frequent for HSV-1. We conclude by proposing a timescale for HSV-1 evolution, and by discussing putative underlying mechanisms for why these otherwise biologically similar viruses have such striking evolutionary differences.
Conflict of interest statement
Figures
References
-
- Roizman B, Sears AE. Herpes simplex viruses and their replication. In: Fields BN, Knipe DM, Howley PM, et al., editors. Virology. Philadelphia: Lippincott-Raven; 1996. pp. 2231–2295.
-
- Lowhagen GB, Tunback P, Bergstrom T. Proportion of herpes simplex virus (HSV) type 1 and type 2 among genital and extragenital HSV isolates. Acta Derm Venereol. 2002;82:118–120. - PubMed
-
- Whitley R, Lakeman AD, Nahmias A, Roizman B. Dna restriction-enzyme analysis of herpes simplex virus isolates obtained from patients with encephalitis. N Engl J Med. 1982;307:1060–1062. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
