

Ehlers-Danlos arthrochalasia type (VIIA-B)--expanding the phenotype: from prenatal life through adulthood
- PMID: 21801164
- PMCID: PMC4026000
- DOI: 10.1111/j.1399-0004.2011.01758.x
Ehlers-Danlos arthrochalasia type (VIIA-B)--expanding the phenotype: from prenatal life through adulthood
Abstract
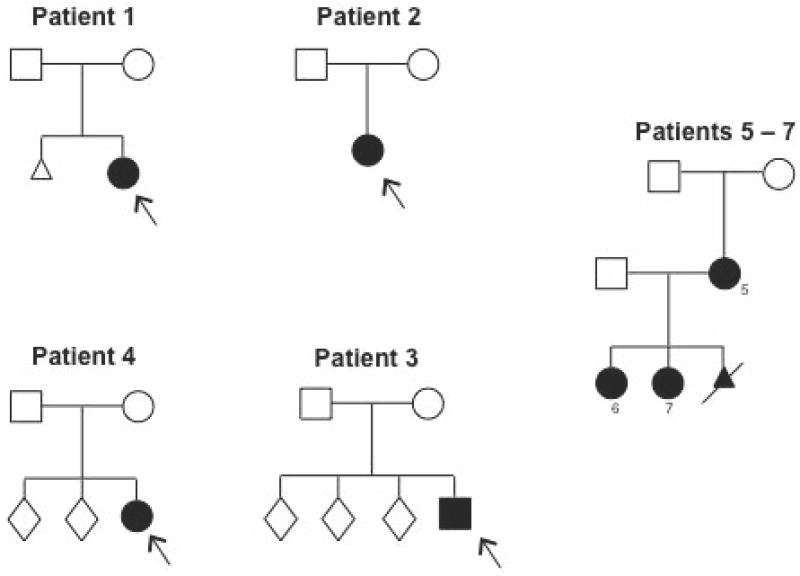
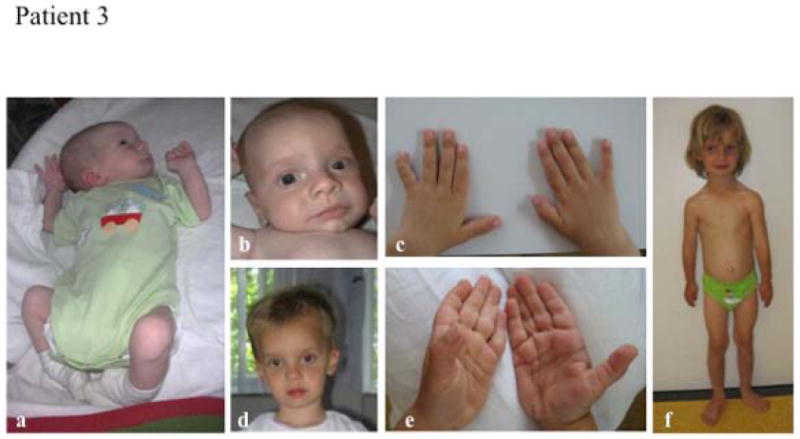
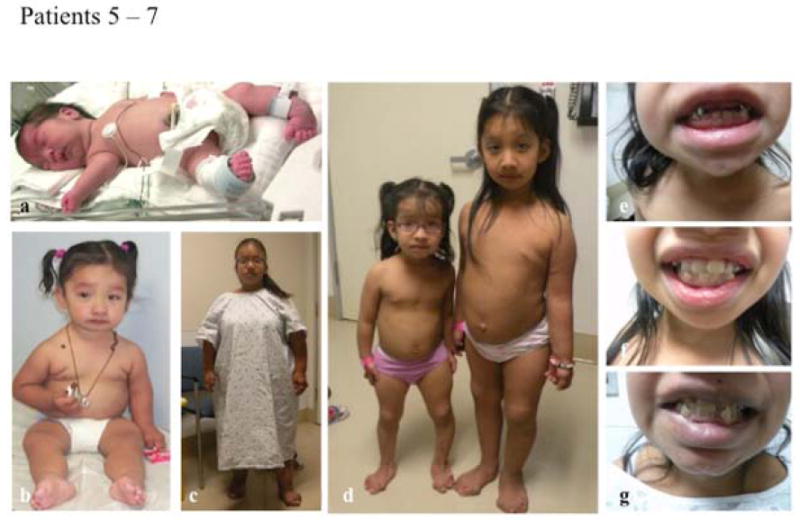
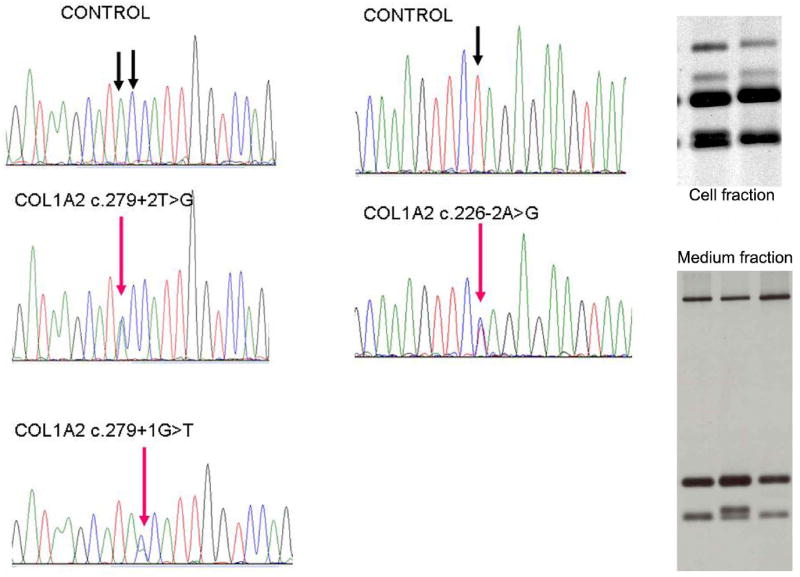
The Ehlers-Danlos syndromes (EDS) form a clinically and genetically heterogeneous group of inherited connective-tissue disorders characterized by joint hypermobility, tissue fragility and skin abnormalities. Six subtypes have been well characterized based on clinical features and molecular genetic abnormalities. The arthrochalasia type EDS (formerly types VIIA and B) is characterized by severe generalized joint hypermobility with multiple dislocations including congenital bilateral dislocation of the hips, muscular hypotonia and distinct dysmorphic features. The diagnosis of the arthrochalasia type EDS is of importance in the neonatal period because of consequences of physical disability in later life. However, the differential diagnosis may be difficult because of overlap with other hypermobility syndromes. In addition, the significant hypotonia may direct the physician toward various neuromuscular diagnoses. As patients become older, the hypotonia decreases and facial features become less distinct. In this report, we describe seven patients at different ages. Timing of diagnosis varied from prenatal life to adult age. The diagnosis of EDS type VII was confirmed by biochemical studies or mutation analysis showing characteristic mutations in COL1A1 and COL1A2. These mutations result in skipping of exon 6, which leads to defective collagen synthesis. For physicians treating patients with EDS type VII, achieving mobility for the patient is the greatest challenge and it may be impossible because of recurrent dislocations of nearly all joints in severe cases.
© 2011 John Wiley & Sons A/S.
Figures



Similar articles
-
Clinical features, molecular results, and management of 12 individuals with the rare arthrochalasia Ehlers-Danlos syndrome.Am J Med Genet A. 2020 May;182(5):994-1007. doi: 10.1002/ajmg.a.61523. Epub 2020 Feb 24. Am J Med Genet A. 2020. PMID: 32091183
-
Pathologic Skull Fracture in a Near-Term Neonate with Arthrochalasia Type Ehlers-Danlos Syndrome: A Case Report.Fetal Pediatr Pathol. 2022 Feb;41(1):149-154. doi: 10.1080/15513815.2020.1753269. Epub 2020 Apr 27. Fetal Pediatr Pathol. 2022. PMID: 32338564
-
The first Japanese case of the arthrochalasia type of Ehlers-Danlos syndrome with COL1A2 gene mutation.Gene. 2014 Mar 15;538(1):199-203. doi: 10.1016/j.gene.2014.01.033. Epub 2014 Jan 17. Gene. 2014. PMID: 24440294
-
Delineation of Ehlers-Danlos syndrome phenotype due to the c.934C>T, p.(Arg312Cys) mutation in COL1A1: Report on a three-generation family without cardiovascular events, and literature review.Am J Med Genet A. 2017 Feb;173(2):524-530. doi: 10.1002/ajmg.a.38035. Epub 2016 Nov 7. Am J Med Genet A. 2017. PMID: 28102596 Review.
-
Mutations in the lysyl hydroxylase 1 gene that result in enzyme deficiency and the clinical phenotype of Ehlers-Danlos syndrome type VI.Mol Genet Metab. 2000 Sep-Oct;71(1-2):212-24. doi: 10.1006/mgme.2000.3076. Mol Genet Metab. 2000. PMID: 11001813 Review.
Cited by
-
Visceroptosis of the bowel in the hypermobility type of Ehlers-Danlos syndrome: presentation of a rare manifestation and review of the literature.Eur J Med Genet. 2012 Oct;55(10):548-51. doi: 10.1016/j.ejmg.2012.06.012. Epub 2012 Jul 7. Eur J Med Genet. 2012. PMID: 22781752 Free PMC article. Review.
-
PITX2 deficiency and associated human disease: insights from the zebrafish model.Hum Mol Genet. 2018 May 15;27(10):1675-1695. doi: 10.1093/hmg/ddy074. Hum Mol Genet. 2018. PMID: 29506241 Free PMC article.
-
A multi-ethnic genome-wide association study implicates collagen matrix integrity and cell differentiation pathways in keratoconus.Commun Biol. 2021 Mar 1;4(1):266. doi: 10.1038/s42003-021-01784-0. Commun Biol. 2021. PMID: 33649486 Free PMC article.
-
Ehlers-Danlos syndromes and their manifestations in the visual system.Front Med (Lausanne). 2022 Sep 27;9:996458. doi: 10.3389/fmed.2022.996458. eCollection 2022. Front Med (Lausanne). 2022. PMID: 36237549 Free PMC article. Review.
-
Helical mutations in type I collagen that affect the processing of the amino-propeptide result in an Osteogenesis Imperfecta/Ehlers-Danlos Syndrome overlap syndrome.Orphanet J Rare Dis. 2013 May 21;8:78. doi: 10.1186/1750-1172-8-78. Orphanet J Rare Dis. 2013. PMID: 23692737 Free PMC article.
References
-
- Beighton P, De Paepe A, Steinmann B, et al. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am J Med Genet. 1998;77:31–37. - PubMed
-
- Yen JL, Lin SP, Chen MR, et al. Clinical features of Ehlers-Danlos syndrome. J Formos Med Assoc. 2006;105:475–480. - PubMed
-
- Hass J, Hass R. Arthrochalasis multiplex congenita; congenital flaccidity of the joints. J Bone Joint Surg Am. 1958;40-A:663–674. - PubMed
-
- Giunta C, Superti-Furga A, Spranger S, et al. Ehlers-Danlos syndrome type VII: clinical features and molecular defects. J Bone Joint Surg Am. 1999;81:225–238. - PubMed
-
- Byers PH, Duvic M, Atkinson M, et al. Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet. 1997;72:94–105. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
