

Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma
- PMID: 21803744
- PMCID: PMC3174331
- DOI: 10.1158/0008-5472.CAN-11-0595
Activated Notch1 induces lung adenomas in mice and cooperates with Myc in the generation of lung adenocarcinoma
Abstract
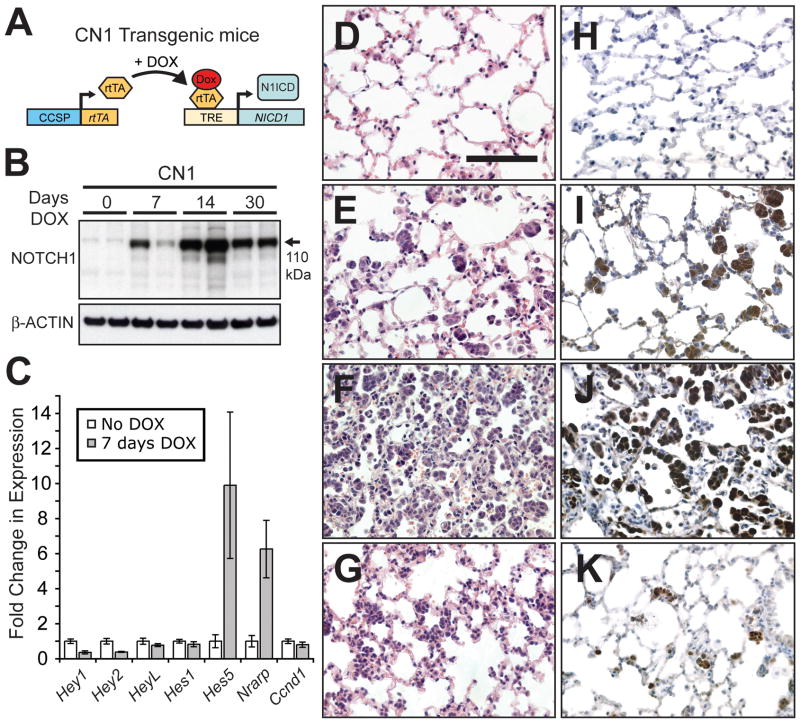
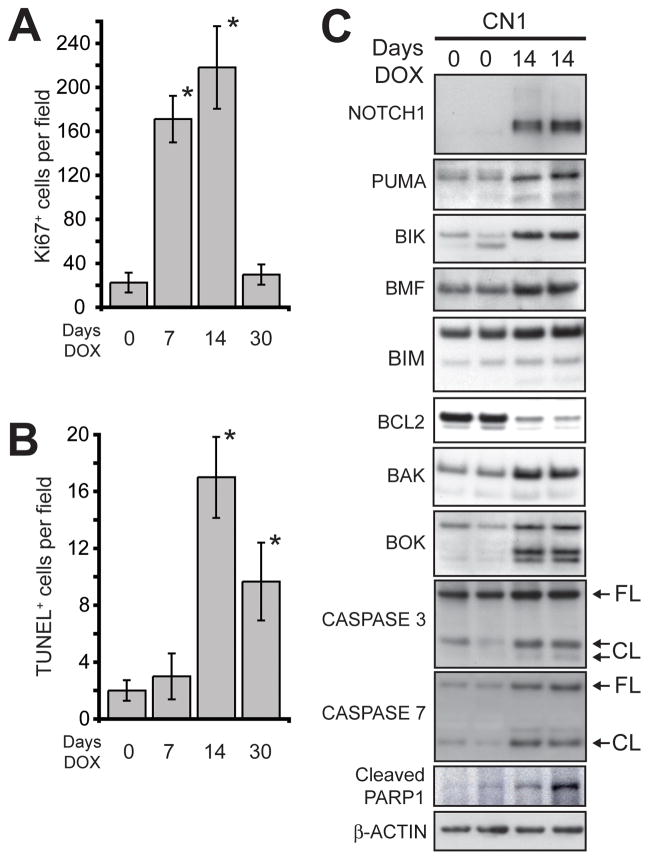
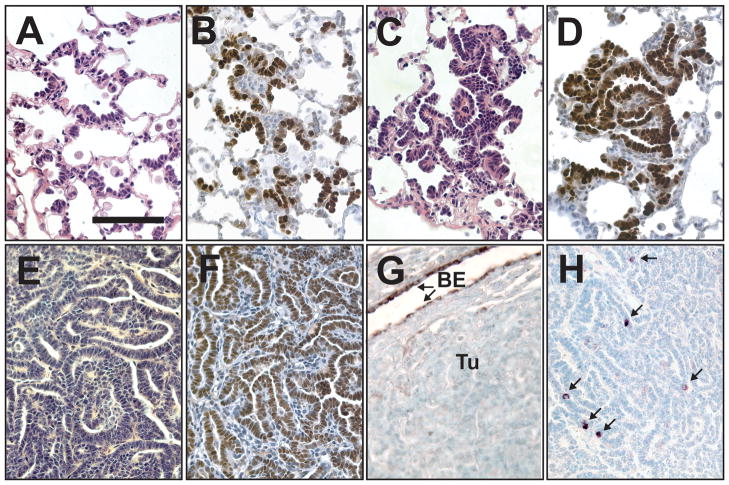
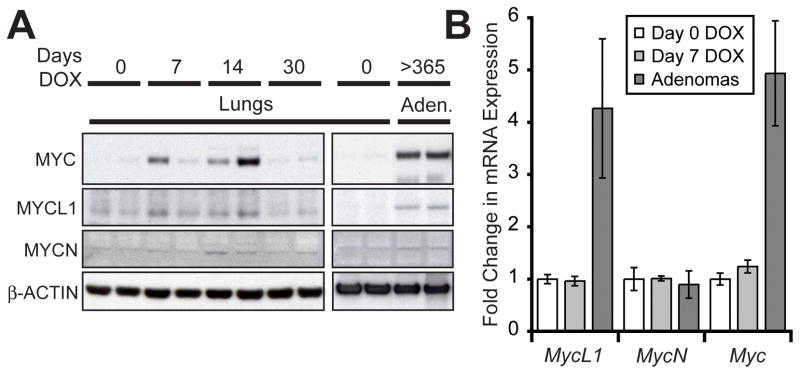
Notch1 encodes the canonical member of the mammalian Notch receptor family. Activating lesions frequently affect Notch1 in T-cell acute lymphoblastic leukemia (T-ALL) and, recently, have been found in non-small-cell lung cancer (NSCLC) as well. We explored the oncogenic potential of activated Notch1 in the lung by developing a transgenic mouse model in which activated Notch1 was overexpressed in the alveolar epithelium. The initial response to activated Notch1 was proliferation and the accumulation of alveolar hyperplasia, which was then promptly cleared by apoptosis. After an extended latency period, however, pulmonary adenomas appeared in the transgenic mice but failed to progress to become carcinomas. Interestingly, Myc and MycL1 were expressed in the adenomas, suggesting that selection for enhanced Myc activity may facilitate tumorigenesis. Using mice engineered to coexpress activated Notch1 and Myc, we found that supplementing Myc expression resulted in increased frequency of Notch1 intracellular domain (N1ICD)-induced adenoma formation and enabled progression to adenocarcinoma and metastases. Cooperation stemmed from synergistic activation of tumor cell cycling, a process that apparently countered any impedance to tumorigenesis posed by Myc and/or activated Notch1-induced apoptosis. Significantly, cooperation was independent of RAS activation. Taken together, the data suggest that activated Notch1 substitutes for RAS activation synergistically with Myc in the development of NSCLC. These tumor models should be valuable for exploring the role of activated Notch1 in the genesis of NSCLC and for testing therapies targeting either activated Notch1 or its downstream effectors.
Conflict of interest statement
Conflicts of interest: None
Figures
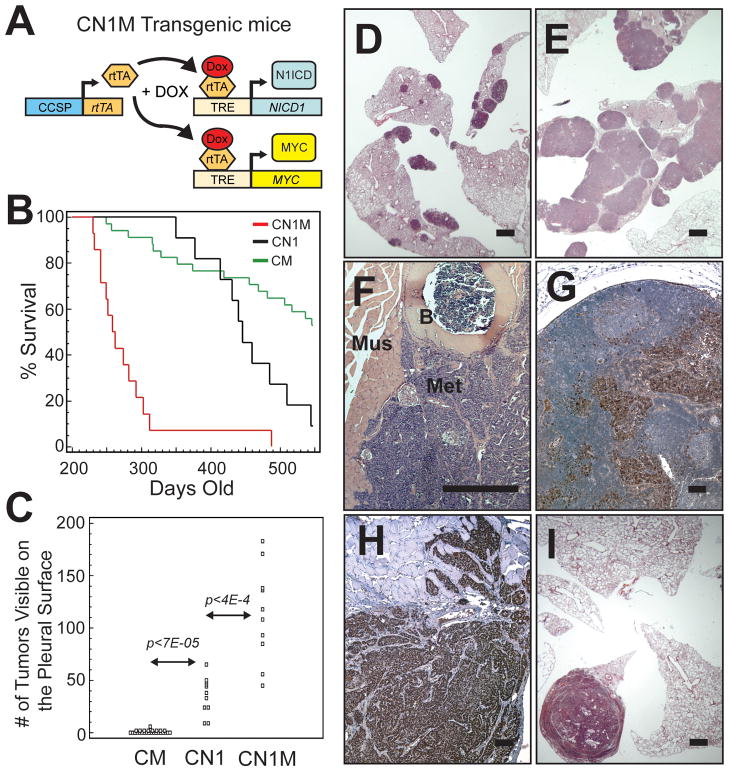
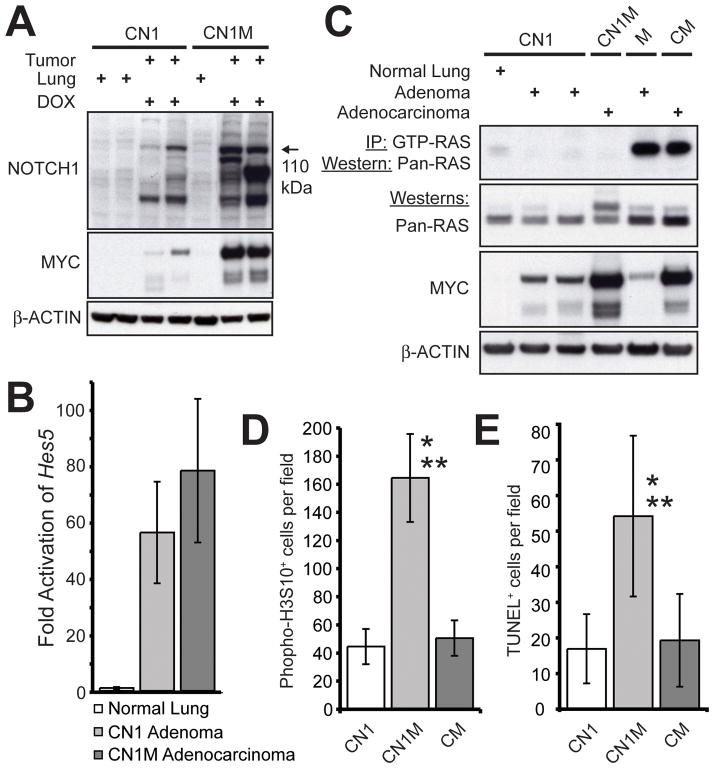
References
-
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. - PubMed
-
- Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature. 1998;393:382–6. - PubMed
-
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–8. - PubMed
-
- Ehebauer M, Hayward P, Martinez-Arias A. Notch signaling pathway. Sci STKE. 2006;2006:cm7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
