

Measuring cell identity in noisy biological systems
- PMID: 21803789
- PMCID: PMC3241637
- DOI: 10.1093/nar/gkr591
Measuring cell identity in noisy biological systems
Abstract
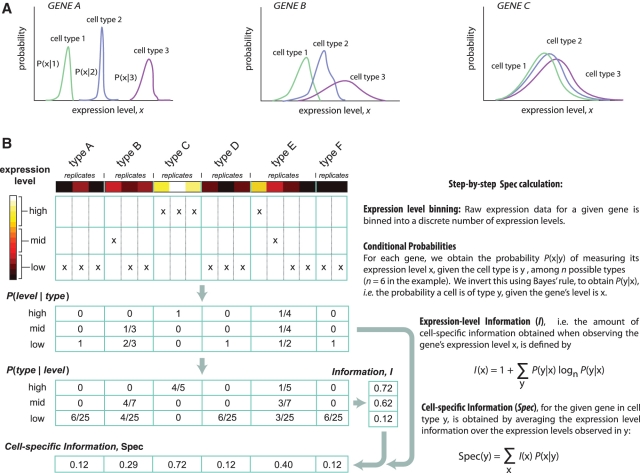
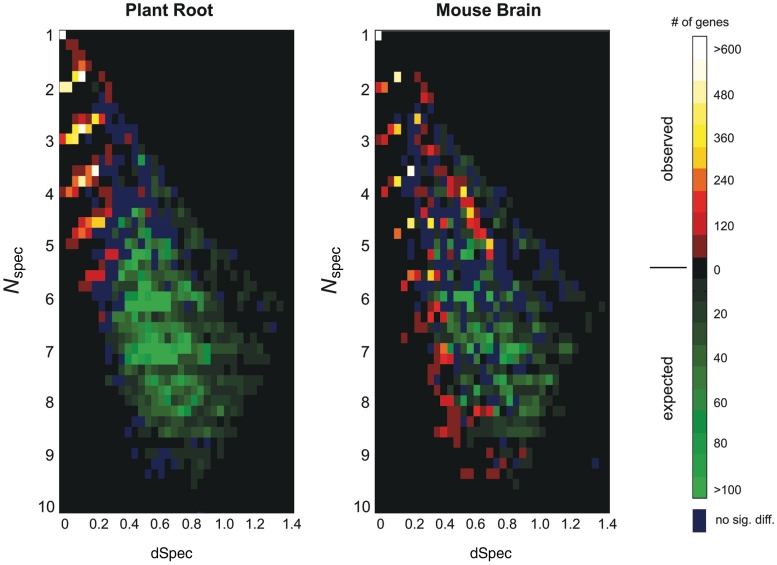
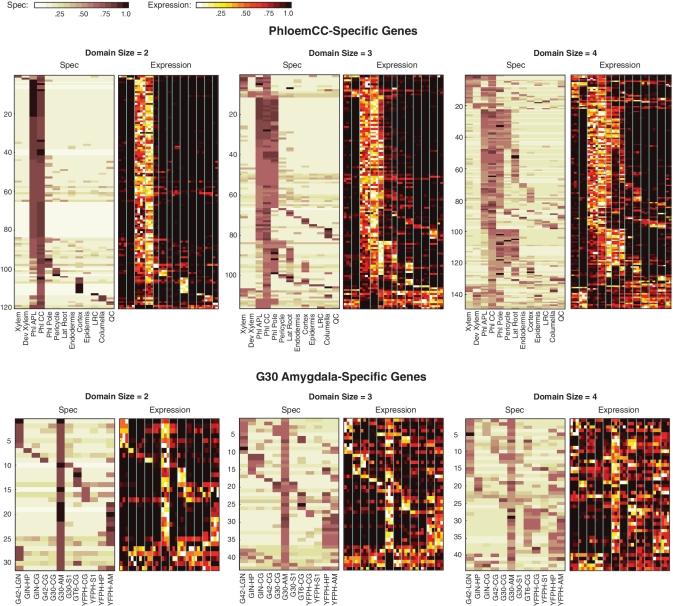
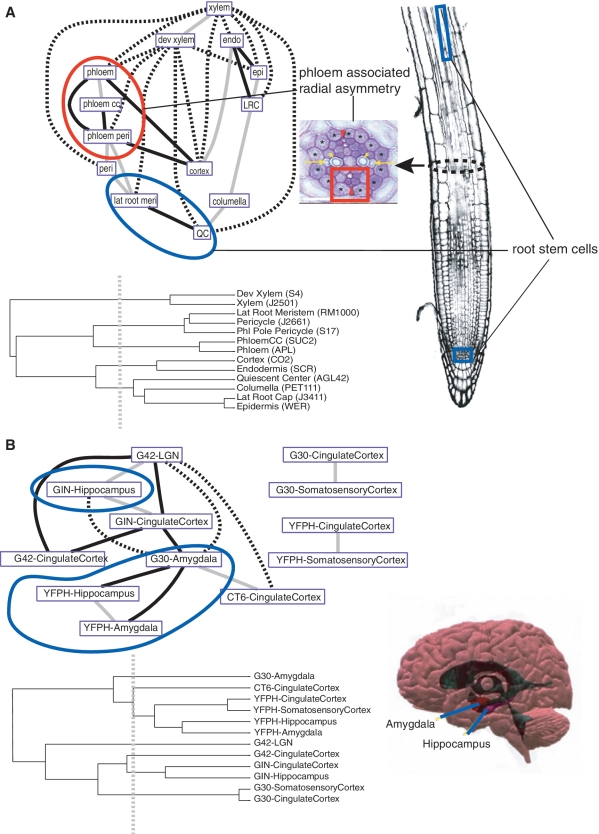
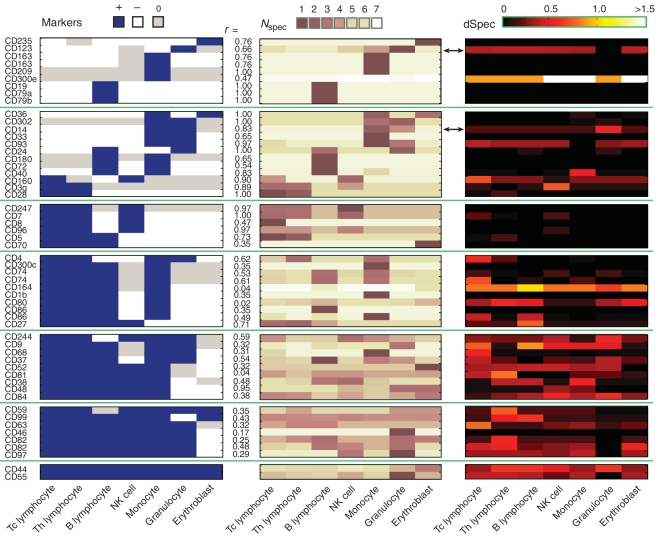
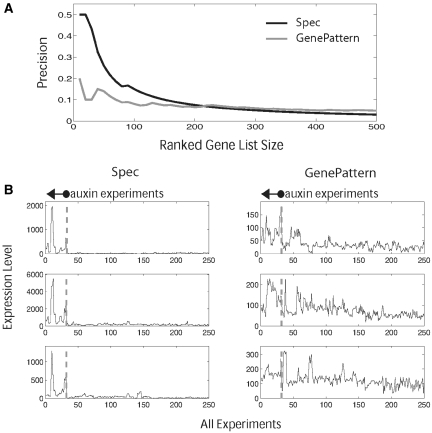
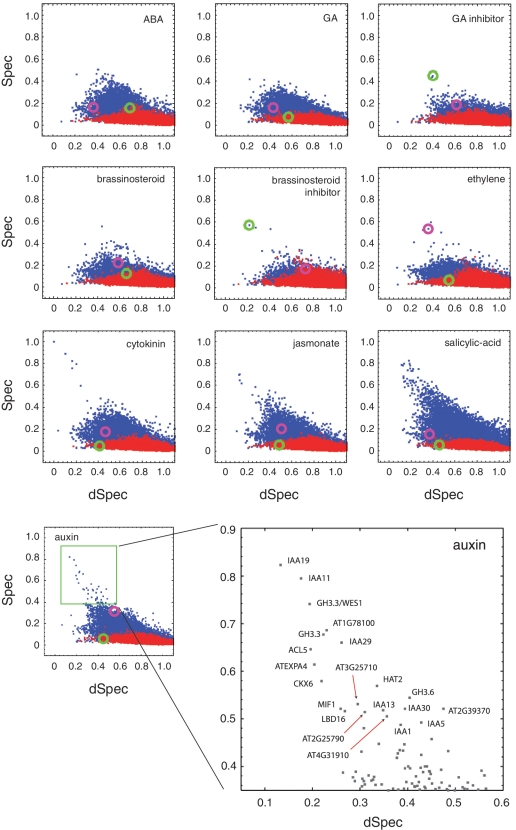
Global gene expression measurements are increasingly obtained as a function of cell type, spatial position within a tissue and other biologically meaningful coordinates. Such data should enable quantitative analysis of the cell-type specificity of gene expression, but such analyses can often be confounded by the presence of noise. We introduce a specificity measure Spec that quantifies the information in a gene's complete expression profile regarding any given cell type, and an uncertainty measure dSpec, which measures the effect of noise on specificity. Using global gene expression data from the mouse brain, plant root and human white blood cells, we show that Spec identifies genes with variable expression levels that are nonetheless highly specific of particular cell types. When samples from different individuals are used, dSpec measures genes' transcriptional plasticity in each cell type. Our approach is broadly applicable to mapped gene expression measurements in stem cell biology, developmental biology, cancer biology and biomarker identification. As an example of such applications, we show that Spec identifies a new class of biomarkers, which exhibit variable expression without compromising specificity. The approach provides a unifying theoretical framework for quantifying specificity in the presence of noise, which is widely applicable across diverse biological systems.
Figures
References
-
- Gould J, Getz G, Monti S, Reich M, Mesirov JP. Comparative gene marker selection suite. Bioinformatics. 2006;22:1924–1925. - PubMed
-
- Arendt D. The evolution of cell types in animals: emerging principles from molecular studies. Nat. Rev. Genet. 2008;9:868–882. - PubMed
-
- Sugino K, Hempel CM, Miller MN, Hattox AM, Shapiro P, Wu C, Huang ZJ, Nelson SB. Molecular taxonomy of major neuronal classes in the adult mouse forebrain. Nat. Neurosci. 2006;9:99–107. - PubMed
-
- Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN. A gene expression map of the Arabidopsis root. Science. 2003;302:1956–1960. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
