

True single-molecule DNA sequencing of a pleistocene horse bone
- PMID: 21803858
- PMCID: PMC3202287
- DOI: 10.1101/gr.122747.111
True single-molecule DNA sequencing of a pleistocene horse bone
Abstract
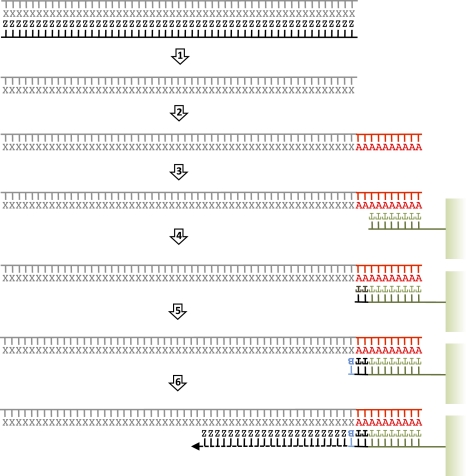
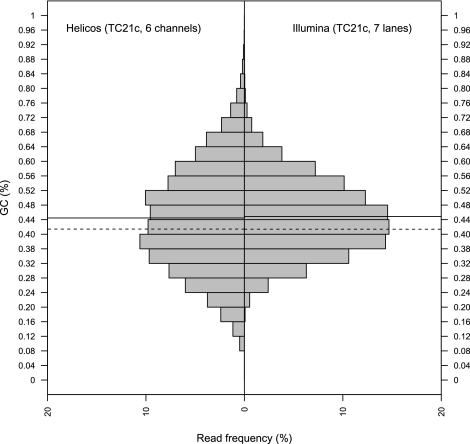
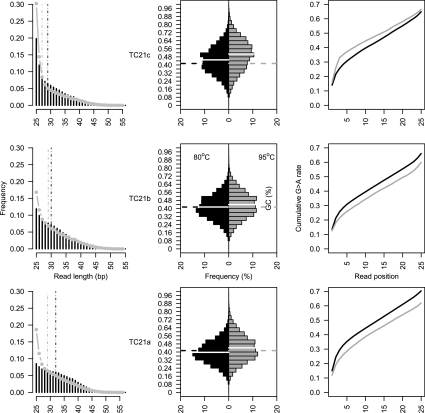
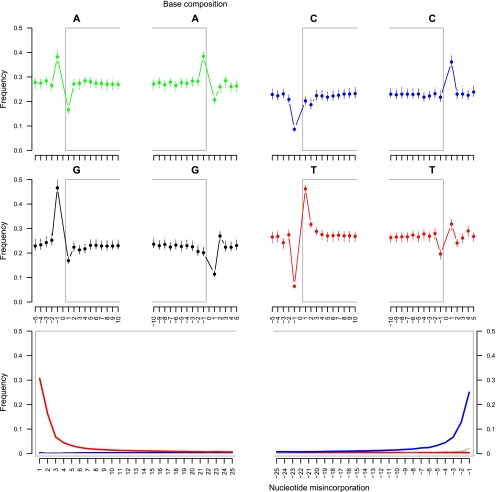
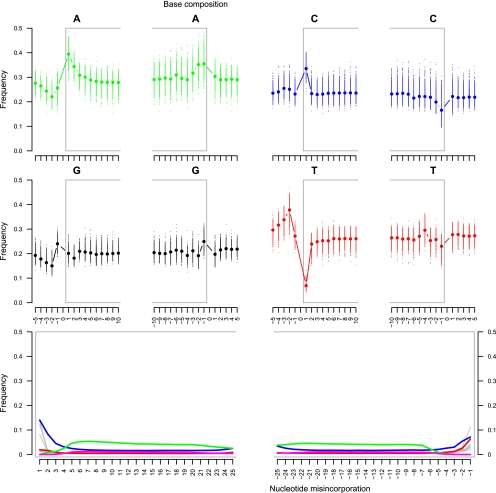
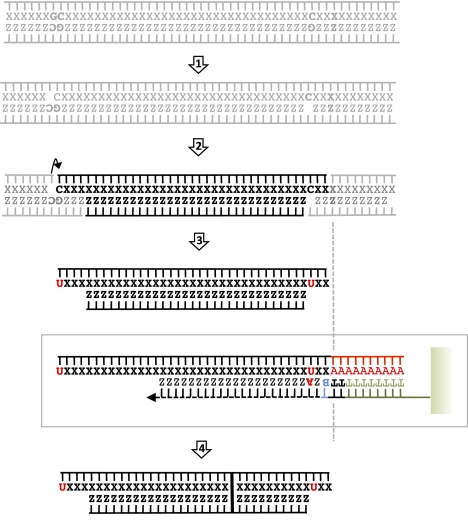
Second-generation sequencing platforms have revolutionized the field of ancient DNA, opening access to complete genomes of past individuals and extinct species. However, these platforms are dependent on library construction and amplification steps that may result in sequences that do not reflect the original DNA template composition. This is particularly true for ancient DNA, where templates have undergone extensive damage post-mortem. Here, we report the results of the first "true single molecule sequencing" of ancient DNA. We generated 115.9 Mb and 76.9 Mb of DNA sequences from a permafrost-preserved Pleistocene horse bone using the Helicos HeliScope and Illumina GAIIx platforms, respectively. We find that the percentage of endogenous DNA sequences derived from the horse is higher among the Helicos data than Illumina data. This result indicates that the molecular biology tools used to generate sequencing libraries of ancient DNA molecules, as required for second-generation sequencing, introduce biases into the data that reduce the efficiency of the sequencing process and limit our ability to fully explore the molecular complexity of ancient DNA extracts. We demonstrate that simple modifications to the standard Helicos DNA template preparation protocol further increase the proportion of horse DNA for this sample by threefold. Comparison of Helicos-specific biases and sequence errors in modern DNA with those in ancient DNA also reveals extensive cytosine deamination damage at the 3' ends of ancient templates, indicating the presence of 3'-sequence overhangs. Our results suggest that paleogenomes could be sequenced in an unprecedented manner by combining current second- and third-generation sequencing approaches.
Figures
References
-
- Bramanti B, Thomas MG, Haak W, Unterlaender M, Jores P, Tambets K, Antanaitis-Jacobs I, Haidle MN, Jankauskas R, Kind C-J, et al. 2009. Genetic discontinuity between local hunter-gatherers and Europe's first farmers. Science 326: 137–140 - PubMed
-
- Briggs AW, Good JM, Green RE, Krause J, Maricic T, Stenzel U, Lalueza-Fox C, Rudan P, Brajkovic D, Kucan Z, et al. 2009. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science 325: 318–321 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous