

Recent advances in the molecular pathophysiology of atrial fibrillation
- PMID: 21804195
- PMCID: PMC3148739
- DOI: 10.1172/JCI46315
Recent advances in the molecular pathophysiology of atrial fibrillation
Abstract
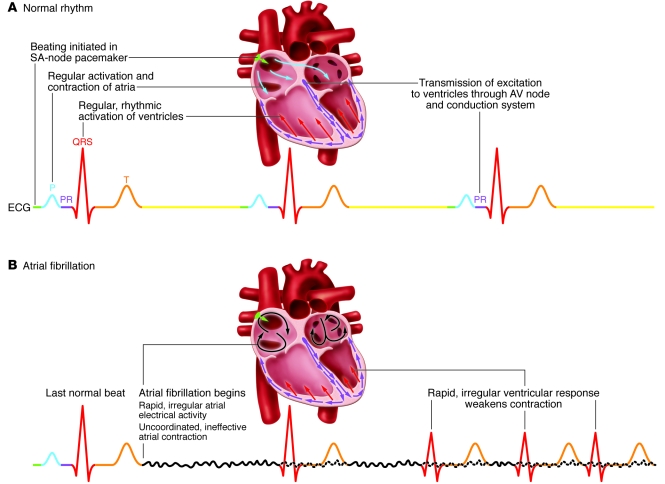
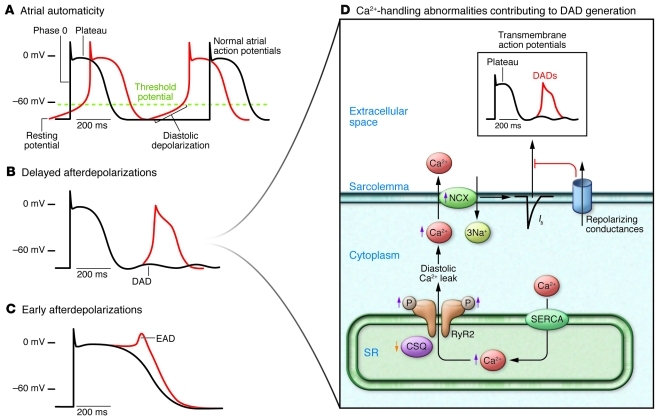
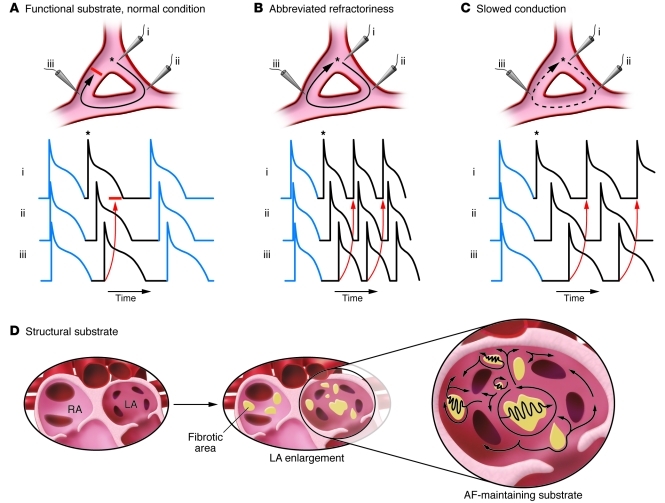
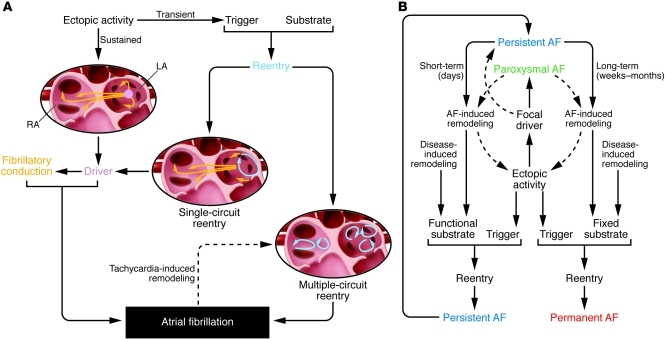
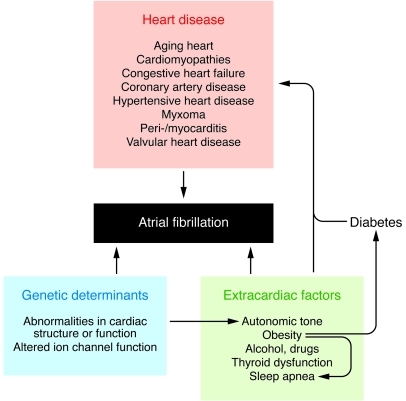
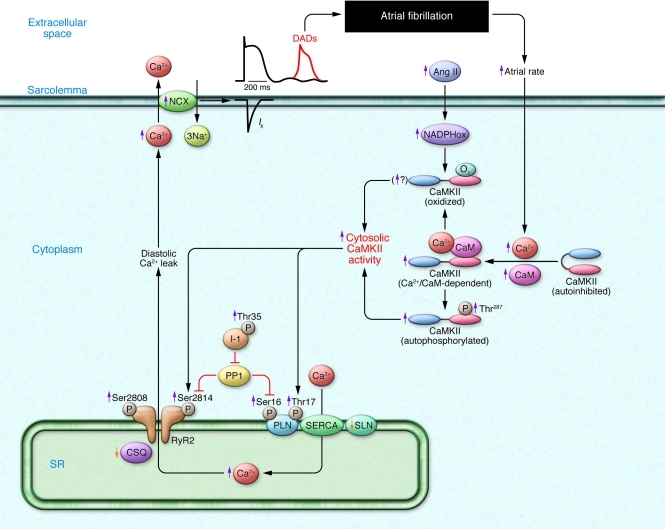
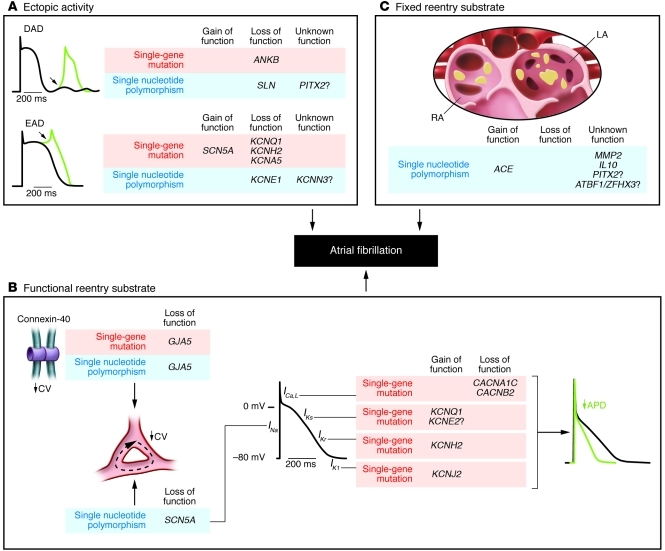
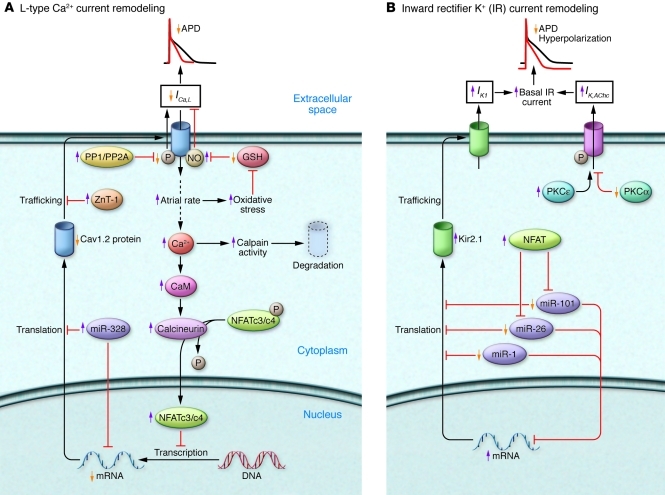
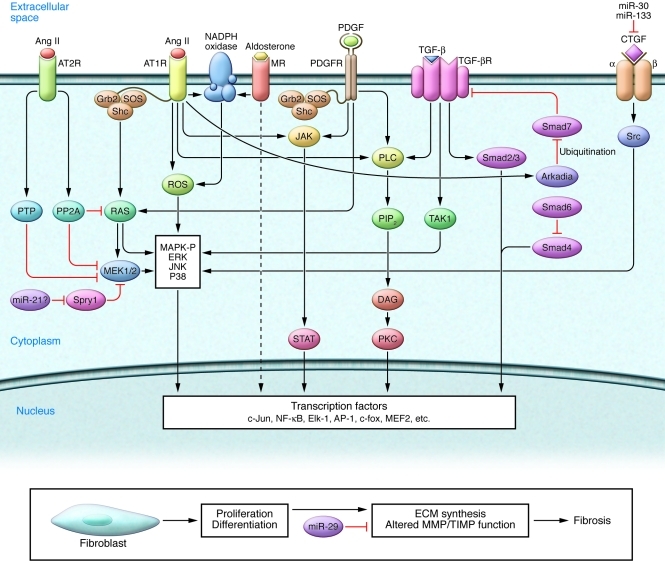
Atrial fibrillation (AF) is an extremely common cardiac rhythm disorder that causes substantial morbidity and contributes to mortality. The mechanisms underlying AF are complex, involving both increased spontaneous ectopic firing of atrial cells and impulse reentry through atrial tissue. Over the past ten years, there has been enormous progress in understanding the underlying molecular pathobiology. This article reviews the basic mechanisms and molecular processes causing AF. We discuss the ways in which cardiac disease states, extracardiac factors, and abnormal genetic control lead to the arrhythmia. We conclude with a discussion of the potential therapeutic implications that might arise from an improved mechanistic understanding.
Figures
References
-
- Dobrev D, Nattel S. New antiarrhythmic drugs for treatment of atrial fibrillation. Lancet. 2010;375(9721):1212–1223. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
