

Disruption of inflammatory signals by cytokine-targeted therapies for inflammatory bowel diseases
- PMID: 21806600
- PMCID: PMC3312480
- DOI: 10.1111/j.1476-5381.2011.01614.x
Disruption of inflammatory signals by cytokine-targeted therapies for inflammatory bowel diseases
Abstract
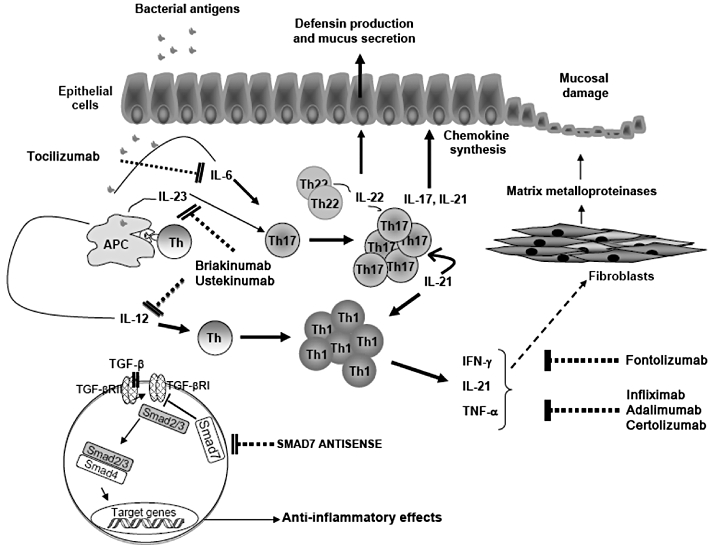
Gut inflammation occurring in patients with inflammatory bowel diseases (IBD) is associated with an excessive immune response that is directed against constituents of the normal bacterial flora and results in the production of large amounts of inflammatory cytokines. Anti-cytokine compounds, such as the neutralizing TNF antibodies, have been employed with clinical success in patients with IBD. However, nearly half of IBD patients are refractory to such treatments, response can wane with time, and anti-TNF treatment can associate with severe side effects and/or development/exacerbation of extra-intestinal immune-mediated pathologies. These observations, and the demonstration that, in IBD, the pathological process is also characterized by defects in the production and/or activity of counter-regulatory cytokines, have boosted further studies aimed at delineating novel strategies to combat the IBD-associated tissue-damaging immune response.
© 2011 The Authors. British Journal of Pharmacology © 2011 The British Pharmacological Society.
Figures
Similar articles
-
Targets for new immunomodulation strategies in inflammatory bowel disease.Autoimmun Rev. 2014 Jan;13(1):11-4. doi: 10.1016/j.autrev.2013.06.003. Epub 2013 Jun 15. Autoimmun Rev. 2014. PMID: 23774108 Review.
-
What's the next best cytokine target in IBD?Inflamm Bowel Dis. 2012 Nov;18(11):2180-9. doi: 10.1002/ibd.22967. Epub 2012 Apr 16. Inflamm Bowel Dis. 2012. PMID: 22508526 Review.
-
Strategies for targeting cytokines in inflammatory bowel disease.Nat Rev Immunol. 2024 Aug;24(8):559-576. doi: 10.1038/s41577-024-01008-6. Epub 2024 Mar 14. Nat Rev Immunol. 2024. PMID: 38486124 Review.
-
A Decade of Th9 Cells: Role of Th9 Cells in Inflammatory Bowel Disease.Front Immunol. 2018 May 24;9:1139. doi: 10.3389/fimmu.2018.01139. eCollection 2018. Front Immunol. 2018. PMID: 29881387 Free PMC article. Review.
-
Biologic therapy for inflammatory bowel disease.Drugs. 2005;65(16):2253-86. doi: 10.2165/00003495-200565160-00002. Drugs. 2005. PMID: 16266194 Review.
Cited by
-
NS6180, a new K(Ca) 3.1 channel inhibitor prevents T-cell activation and inflammation in a rat model of inflammatory bowel disease.Br J Pharmacol. 2013 Jan;168(2):432-44. doi: 10.1111/j.1476-5381.2012.02143.x. Br J Pharmacol. 2013. PMID: 22891655 Free PMC article.
-
Concordance of preclinical and clinical pharmacology and toxicology of monoclonal antibodies and fusion proteins: soluble targets.Br J Pharmacol. 2012 Jun;166(3):806-22. doi: 10.1111/j.1476-5381.2011.01812.x. Br J Pharmacol. 2012. PMID: 22168335 Free PMC article. Review.
-
Inhibition of the inflammatory cytokine tumor necrosis factor-alpha with etanercept provides protection against lethal H1N1 influenza infection in mice.Crit Care. 2013 Dec 27;17(6):R301. doi: 10.1186/cc13171. Crit Care. 2013. PMID: 24373231 Free PMC article.
-
Inhibition of G-Protein βγ Signaling Decreases Levels of Messenger RNAs Encoding Proinflammatory Cytokines in T Cell Receptor-Stimulated CD4(+) T Helper Cells.J Mol Signal. 2015 Jul 6;10:1. doi: 10.5334/1750-2187-10-1. J Mol Signal. 2015. PMID: 27095999 Free PMC article.
References
-
- Anderson P, Louie J, Lau A, Broder M. Mechanisms of differential immunogenicity of tumor necrosis factor inhibitors. Curr Rheumatol Rep. 2005;7:3–9. - PubMed
-
- Arora T, Padaki R, Liu L, Hamburger AE, Ellison AR, Stevens SR, et al. Differences in binding and effector functions between classes of TNF antagonists. Cytokine. 2009;45:124–131. - PubMed
-
- Atreya R, Mudter J, Finotto S, Mullberg J, Jostock T, Wirtz S, et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: evidence in Crohn disease and experimental colitis in vivo. Nat Med. 2000;6:583–588. - PubMed
-
- Atreya R, Atreya I, Neurath MF. Novel signal transduction pathways: analysis of STAT-3 and Rac-1 signaling in inflammatory bowel disease. Ann N Y Acad Sci. 2006;1072:98–113. - PubMed
-
- Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology. 1996;110:975–984. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
