

Inferring causal genomic alterations in breast cancer using gene expression data
- PMID: 21806811
- PMCID: PMC3162519
- DOI: 10.1186/1752-0509-5-121
Inferring causal genomic alterations in breast cancer using gene expression data
Abstract
Background: One of the primary objectives in cancer research is to identify causal genomic alterations, such as somatic copy number variation (CNV) and somatic mutations, during tumor development. Many valuable studies lack genomic data to detect CNV; therefore, methods that are able to infer CNVs from gene expression data would help maximize the value of these studies.
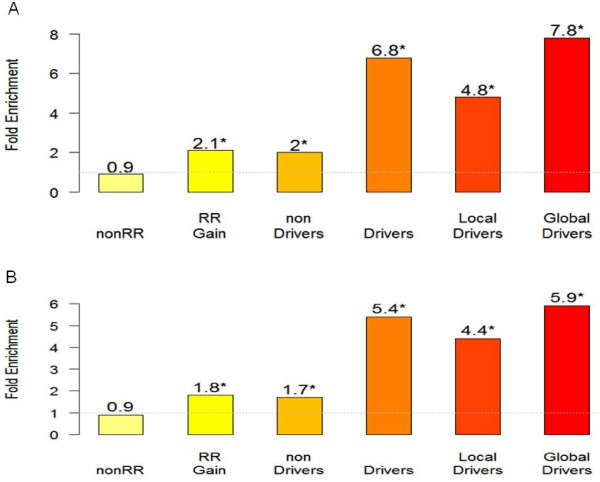
Results: We developed a framework for identifying recurrent regions of CNV and distinguishing the cancer driver genes from the passenger genes in the regions. By inferring CNV regions across many datasets we were able to identify 109 recurrent amplified/deleted CNV regions. Many of these regions are enriched for genes involved in many important processes associated with tumorigenesis and cancer progression. Genes in these recurrent CNV regions were then examined in the context of gene regulatory networks to prioritize putative cancer driver genes. The cancer driver genes uncovered by the framework include not only well-known oncogenes but also a number of novel cancer susceptibility genes validated via siRNA experiments.
Conclusions: To our knowledge, this is the first effort to systematically identify and validate drivers for expression based CNV regions in breast cancer. The framework where the wavelet analysis of copy number alteration based on expression coupled with the gene regulatory network analysis, provides a blueprint for leveraging genomic data to identify key regulatory components and gene targets. This integrative approach can be applied to many other large-scale gene expression studies and other novel types of cancer data such as next-generation sequencing based expression (RNA-Seq) as well as CNV data.
Figures





Similar articles
-
MutComFocal: an integrative approach to identifying recurrent and focal genomic alterations in tumor samples.BMC Syst Biol. 2013 Mar 25;7:25. doi: 10.1186/1752-0509-7-25. BMC Syst Biol. 2013. PMID: 23531283 Free PMC article.
-
Identification of candidate cancer drivers by integrative Epi-DNA and Gene Expression (iEDGE) data analysis.Sci Rep. 2019 Nov 15;9(1):16904. doi: 10.1038/s41598-019-52886-z. Sci Rep. 2019. PMID: 31729402 Free PMC article.
-
The application of gene co-expression network reconstruction based on CNVs and gene expression microarray data in breast cancer.Mol Biol Rep. 2012 Feb;39(2):1627-37. doi: 10.1007/s11033-011-0902-3. Epub 2011 May 25. Mol Biol Rep. 2012. PMID: 21611746
-
Genome-wide copy number analysis in primary breast cancer.Expert Opin Ther Targets. 2012 Mar;16 Suppl 1:S31-5. doi: 10.1517/14728222.2011.636739. Epub 2012 Feb 8. Expert Opin Ther Targets. 2012. PMID: 22313367 Review.
-
Exome sequence read depth methods for identifying copy number changes.Brief Bioinform. 2015 May;16(3):380-92. doi: 10.1093/bib/bbu027. Epub 2014 Aug 28. Brief Bioinform. 2015. PMID: 25169955 Review.
Cited by
-
Decrease of mRNA Editing after Spinal Cord Injury is Caused by Down-regulation of ADAR2 that is Triggered by Inflammatory Response.Sci Rep. 2015 Jul 30;5:12615. doi: 10.1038/srep12615. Sci Rep. 2015. PMID: 26223940 Free PMC article.
-
Applying Expression Profile Similarity for Discovery of Patient-Specific Functional Mutations.High Throughput. 2018 Feb 22;7(1):6. doi: 10.3390/ht7010006. High Throughput. 2018. PMID: 29485617 Free PMC article.
-
Identifying high-risk multiple myeloma patients: A novel approach using a clonal gene signature.Int J Cancer. 2024 Nov 1;155(9):1684-1695. doi: 10.1002/ijc.35057. Epub 2024 Jun 14. Int J Cancer. 2024. PMID: 38874435
-
Reverse engineering biomolecular systems using -omic data: challenges, progress and opportunities.Brief Bioinform. 2012 Jul;13(4):430-45. doi: 10.1093/bib/bbs026. Brief Bioinform. 2012. PMID: 22833495 Free PMC article.
-
Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers.Mol Syst Biol. 2012 Jul 17;8:594. doi: 10.1038/msb.2012.24. Mol Syst Biol. 2012. PMID: 22806142 Free PMC article.
References
-
- Mullighan CG, Downing JR. Genome-wide profiling of genetic alterations in acute lymphoblastic leukemia: recent insights and future directions. Leukemia. 2009. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
