

Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants
- PMID: 21807635
- PMCID: PMC3184252
- DOI: 10.1158/1078-0432.CCR-11-0728
Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants
Abstract
Purpose: Abiraterone is a potent inhibitor of the steroidogenic enzyme CYP17A1 and suppresses tumor growth in patients with castration-resistant prostate cancer (CRPC). The effectiveness of abiraterone in reducing tumor androgens is not known, nor have mechanisms contributing to abiraterone resistance been established.
Experimental design: We treated human CRPC xenografts with abiraterone and measured tumor growth, tissue androgens, androgen receptor (AR) levels, and steroidogenic gene expression versus controls.
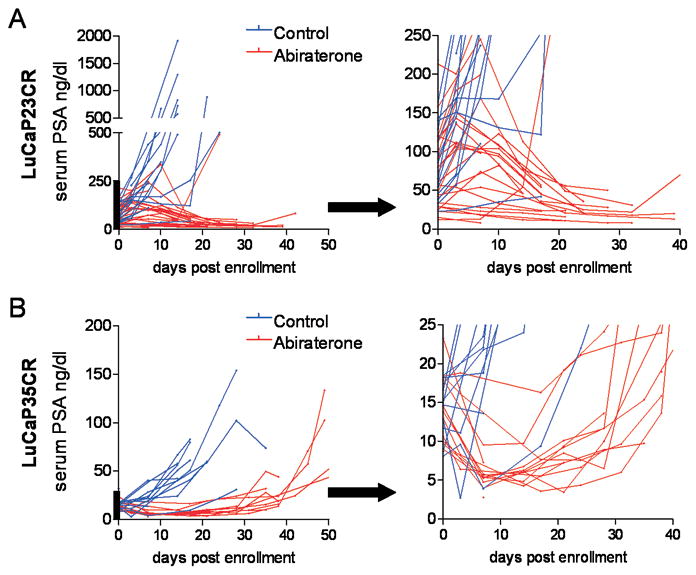
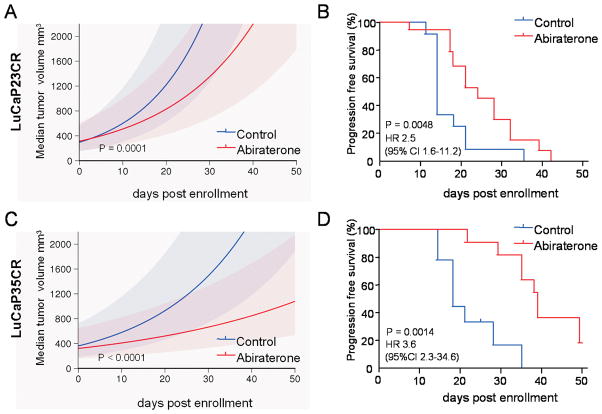
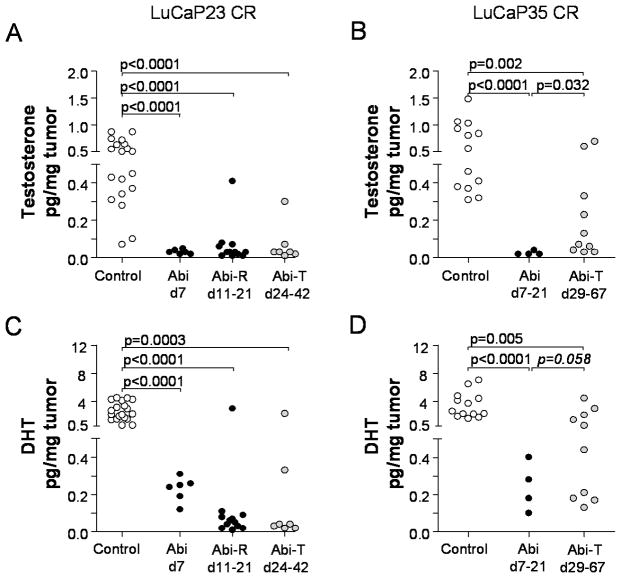
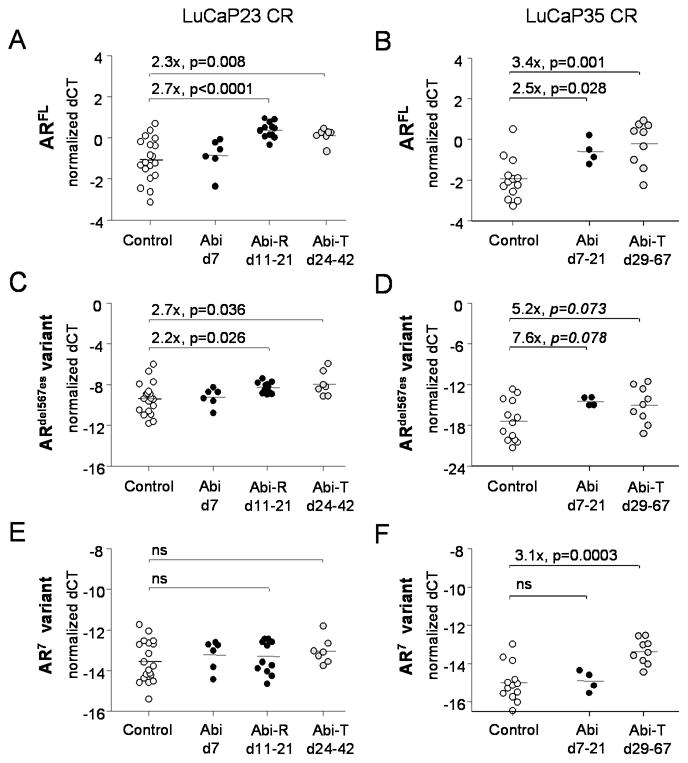
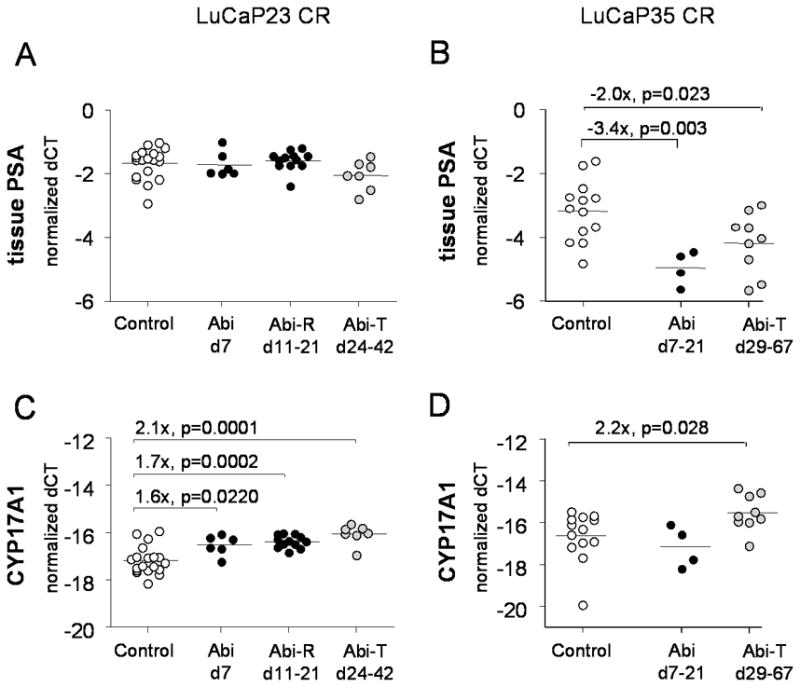
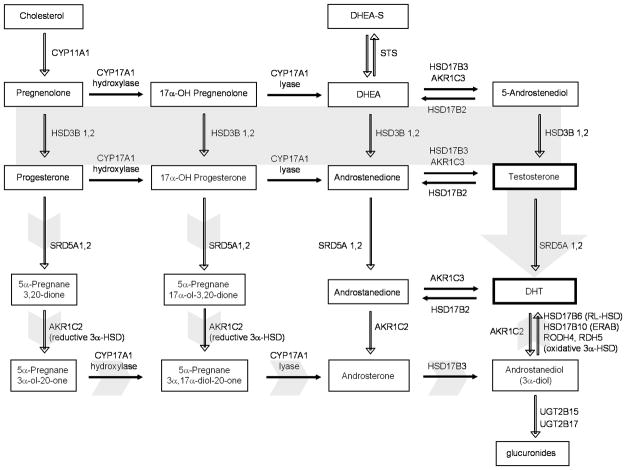
Results: Abiraterone suppressed serum PSA levels and improved survival in two distinct CRPC xenografts: median survival of LuCaP35CR improved from 17 to 39 days (HR = 3.6, P = 0.0014) and LuCaP23CR from 14 to 24 days (HR = 2.5, P = 0.0048). Abiraterone strongly suppressed tumor androgens, with testosterone (T) decreasing from 0.49 ± 0.22 to 0.03 ± 0.01 pg/mg (P < 0.0001), and from 0.69 ± 0.36 to 0.03 ± 0.01 pg/mg (P = 0.002) in abiraterone-treated 23CR and 35CR, respectively, with comparable decreases in tissue DHT. Treatment was associated with increased expression of full-length AR (AR(FL)) and truncated AR variants (AR(FL) 2.3-fold, P = 0.008 and AR(del567es) 2.7-fold, P = 0.036 in 23 CR; AR(FL) 3.4-fold, P = 0.001 and AR(V7) 3.1-fold, P = 0.0003 in 35CR), and increased expression of the abiraterone target CYP17A1 (∼2.1-fold, P = 0.0001 and P = 0.028 in 23CR and 35CR, respectively) and transcript changes in other enzymes modulating steroid metabolism.
Conclusions: These studies indicate that abiraterone reduces CRPC growth via suppression of intratumoral androgens and that resistance to abiraterone may occur through mechanisms that include upregulation of CYP17A1, and/or induction of AR and AR splice variants that confer ligand-independent AR transactivation.
©2011 AACR.
Figures
References
-
- Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, et al. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. - PubMed
-
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. - PubMed
-
- Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. - PubMed
-
- Mostaghel EA, Page ST, Lin DW, Fazli L, Coleman IM, True LD, et al. Intra-prostatic Androgens and Androgen-Regulated Gene Expression Persist after Testosterone Suppression: Therapeutic Implications for Castration-Resistant Prostate Cancer. Cancer Res. 2007;67:5033–41. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
