

Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli
- PMID: 21808005
- PMCID: PMC3158223
- DOI: 10.1073/pnas.1104681108
Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli
Abstract
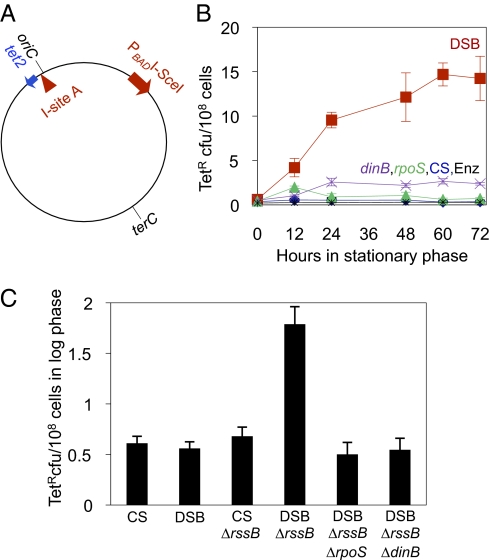
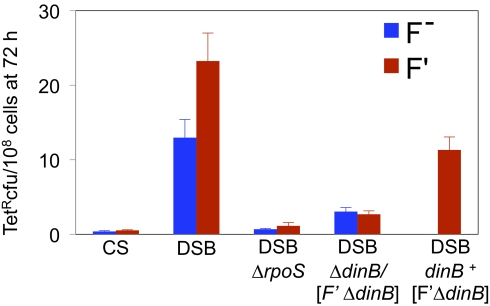
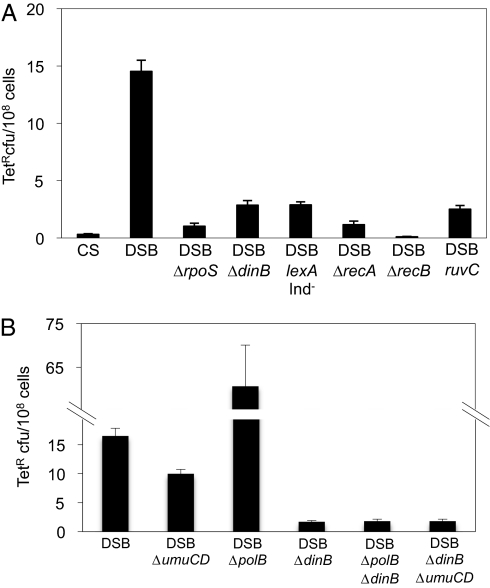
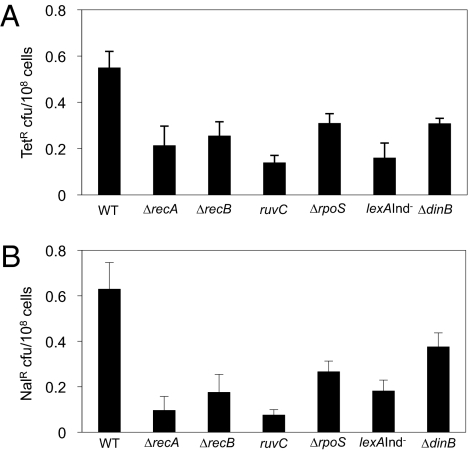
Basic ideas about the constancy and randomness of mutagenesis that drives evolution were challenged by the discovery of mutation pathways activated by stress responses. These pathways could promote evolution specifically when cells are maladapted to their environment (i.e., are stressed). However, the clearest example--a general stress-response-controlled switch to error-prone DNA break (double-strand break, DSB) repair--was suggested to be peculiar to an Escherichia coli F' conjugative plasmid, not generally significant, and to occur by an alternative stress-independent mechanism. Moreover, mechanisms of spontaneous mutation in E. coli remain obscure. First, we demonstrate that this same mechanism occurs in chromosomes of starving F(-) E. coli. I-SceI endonuclease-induced chromosomal DSBs increase mutation 50-fold, dependent upon general/starvation- and DNA-damage-stress responses, DinB error-prone DNA polymerase, and DSB-repair proteins. Second, DSB repair is also mutagenic if the RpoS general-stress-response activator is expressed in unstressed cells, illustrating a stress-response-controlled switch to mutagenic repair. Third, DSB survival is not improved by RpoS or DinB, indicating that mutagenesis is not an inescapable byproduct of repair. Importantly, fourth, fully half of spontaneous frame-shift and base-substitution mutation during starvation also requires the same stress-response, DSB-repair, and DinB proteins. These data indicate that DSB-repair-dependent stress-induced mutation, driven by spontaneous DNA breaks, is a pathway that cells usually use and a major source of spontaneous mutation. These data also rule out major alternative models for the mechanism. Mechanisms that couple mutagenesis to stress responses can allow cells to evolve rapidly and responsively to their environment.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Mayr E. The Growth of Biological Thought: Diversity, Evolution, and Inheritance. The Growth of Biological Thought: Diversity, Evolution, and Inheritance. Cambridge, MA: Harvard University Press; 1985.
-
- Andersson DI, Koskiniemi S, Hughes D. Biological roles of translesion synthesis DNA polymerases in eubacteria. Mol Microbiol. 2010;77:540–548. - PubMed
-
- Radman M. SOS repair hypothesis: Phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci. 1975;5A:355–367. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
