

Structural conservation of druggable hot spots in protein-protein interfaces
- PMID: 21808046
- PMCID: PMC3158149
- DOI: 10.1073/pnas.1101835108
Structural conservation of druggable hot spots in protein-protein interfaces
Abstract
Despite the growing number of examples of small-molecule inhibitors that disrupt protein-protein interactions (PPIs), the origin of druggability of such targets is poorly understood. To identify druggable sites in protein-protein interfaces we combine computational solvent mapping, which explores the protein surface using a variety of small "probe" molecules, with a conformer generator to account for side-chain flexibility. Applications to unliganded structures of 15 PPI target proteins show that the druggable sites comprise a cluster of binding hot spots, distinguishable from other regions of the protein due to their concave topology combined with a pattern of hydrophobic and polar functionality. This combination of properties confers on the hot spots a tendency to bind organic species possessing some polar groups decorating largely hydrophobic scaffolds. Thus, druggable sites at PPI are not simply sites that are complementary to particular organic functionality, but rather possess a general tendency to bind organic compounds with a variety of structures, including key side chains of the partner protein. Results also highlight the importance of conformational adaptivity at the binding site to allow the hot spots to expand to accommodate a ligand of drug-like dimensions. The critical components of this adaptivity are largely local, involving primarily low energy side-chain motions within 6 Å of a hot spot. The structural and physicochemical signature of druggable sites at PPI interfaces is sufficiently robust to be detectable from the structure of the unliganded protein, even when substantial conformational adaptation is required for optimal ligand binding.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
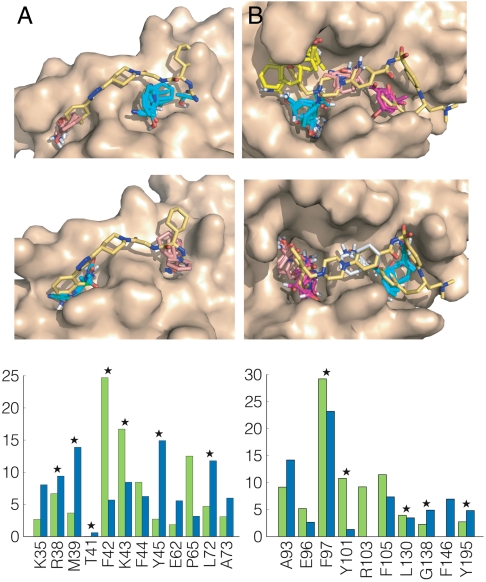
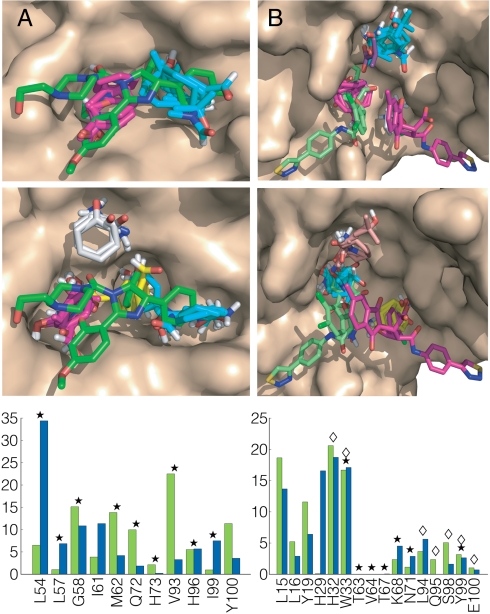
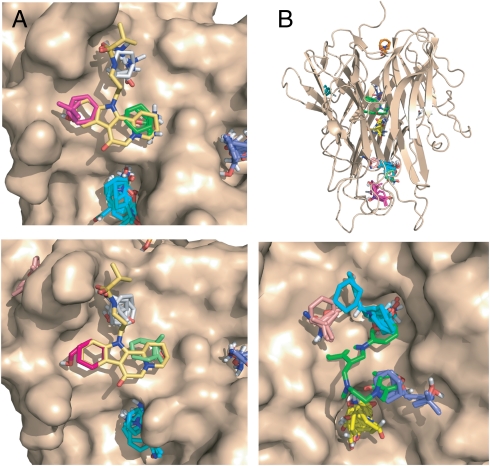
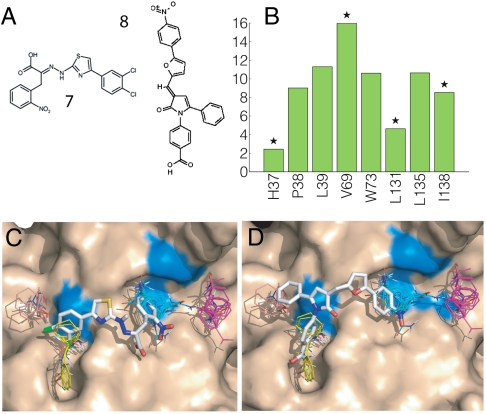
References
-
- Wells J, McClendon C. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. - PubMed
-
- Whitty A, Kumaravel G. Between a rock and a hard place? Nat Chem Biol. 2006;2:112–118. - PubMed
-
- Berg T. Small-molecule inhibitors of protein-protein interactions. Curr Opin Drug Discov Devel. 2008;11:666–674. - PubMed
-
- Fuller JC, Burgoyne NJ, Jackson RM. Predicting druggable binding sites at the protein-protein interface. Drug Disc Today. 2009;14:155–161. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
