

Experimental investigation on the mechanism of chelation-assisted, copper(II) acetate-accelerated azide-alkyne cycloaddition
- PMID: 21809811
- PMCID: PMC3164943
- DOI: 10.1021/ja203733q
Experimental investigation on the mechanism of chelation-assisted, copper(II) acetate-accelerated azide-alkyne cycloaddition
Abstract
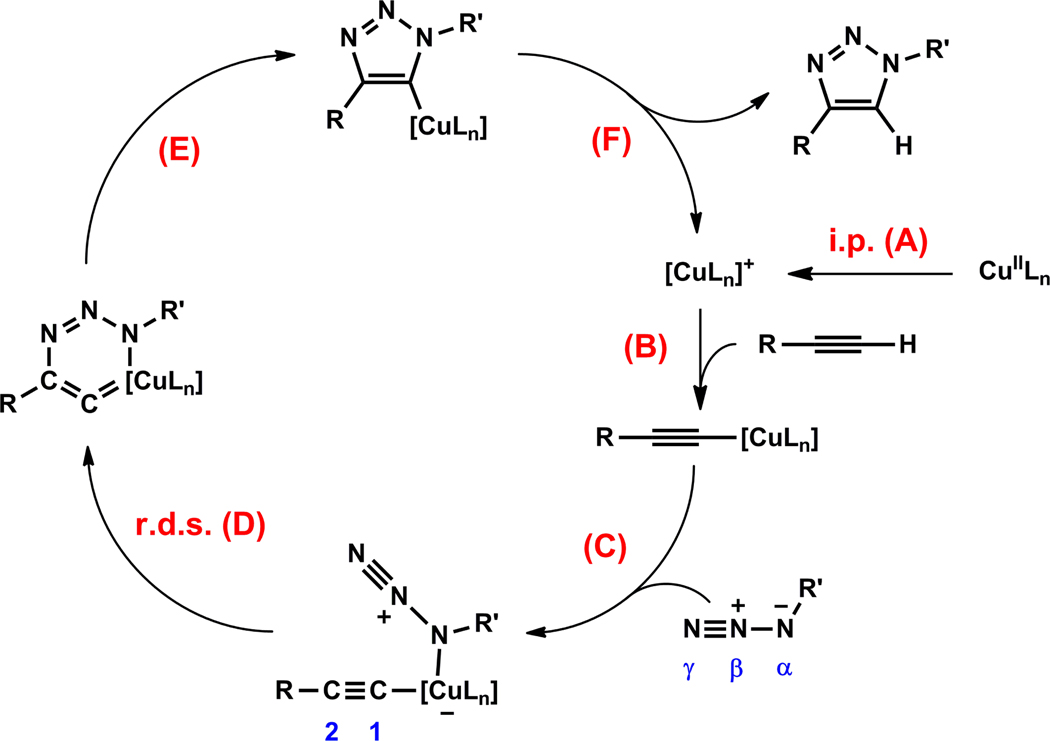
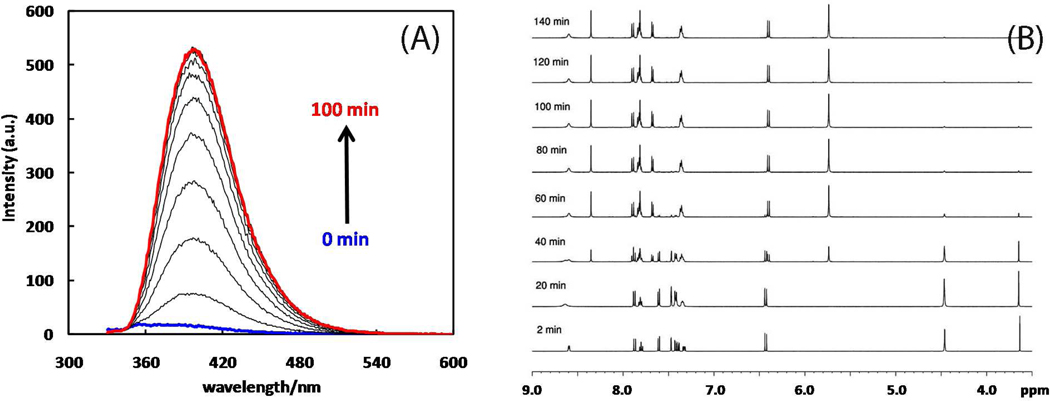
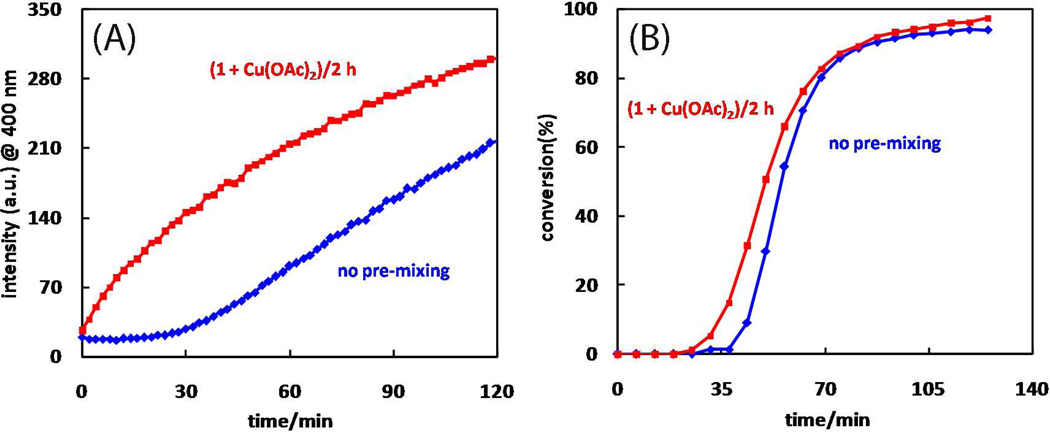
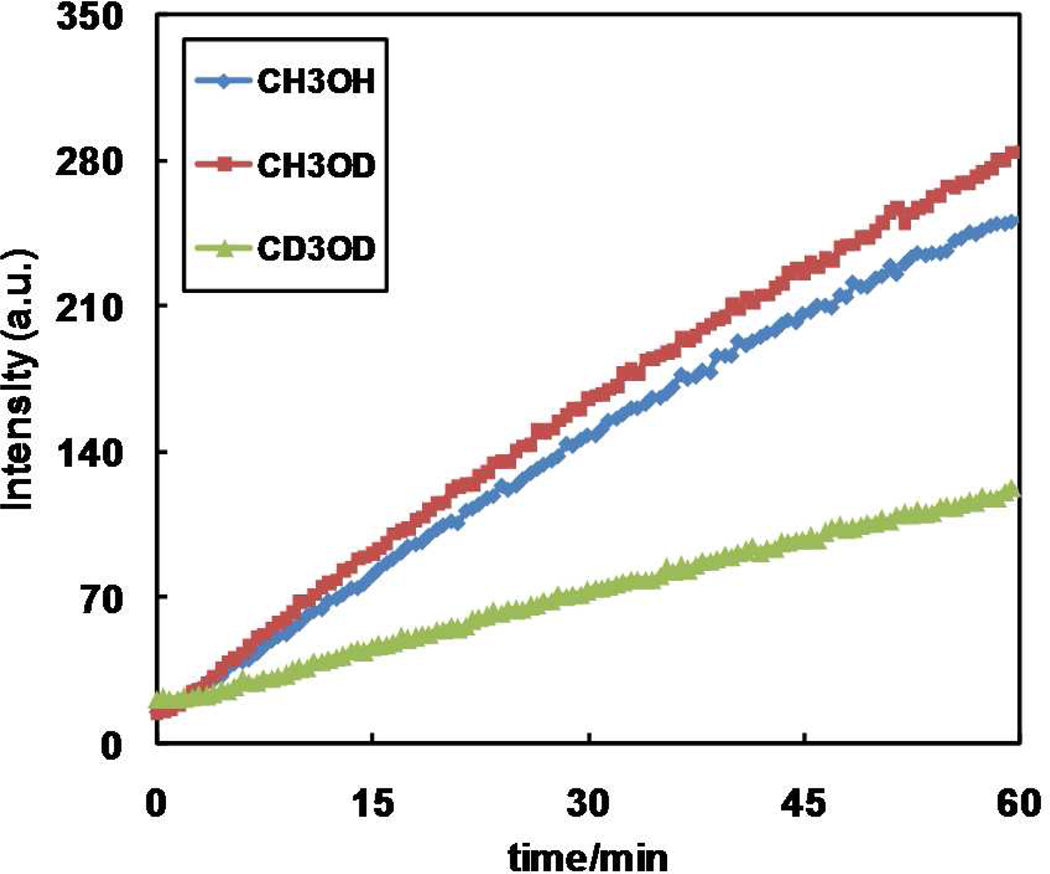
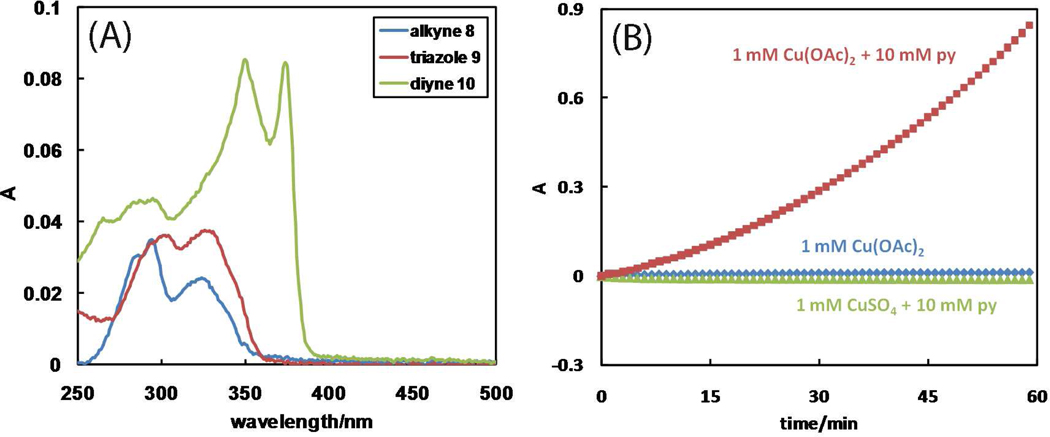
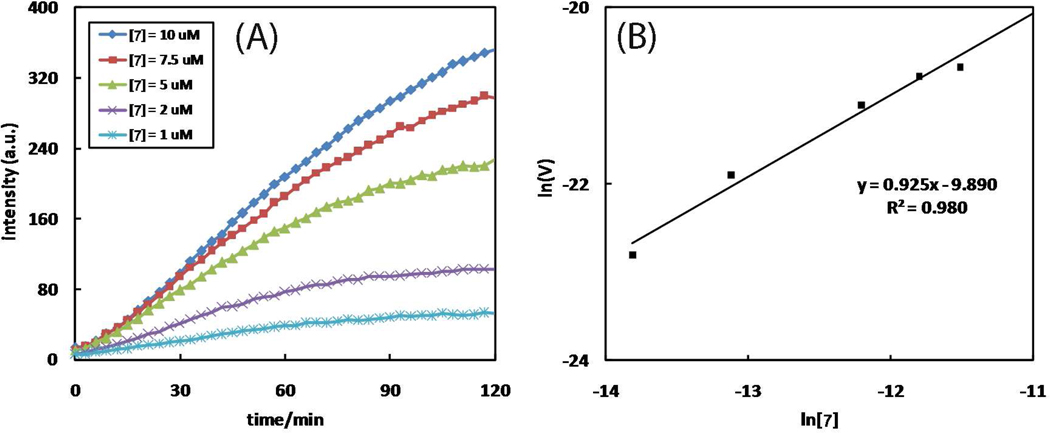
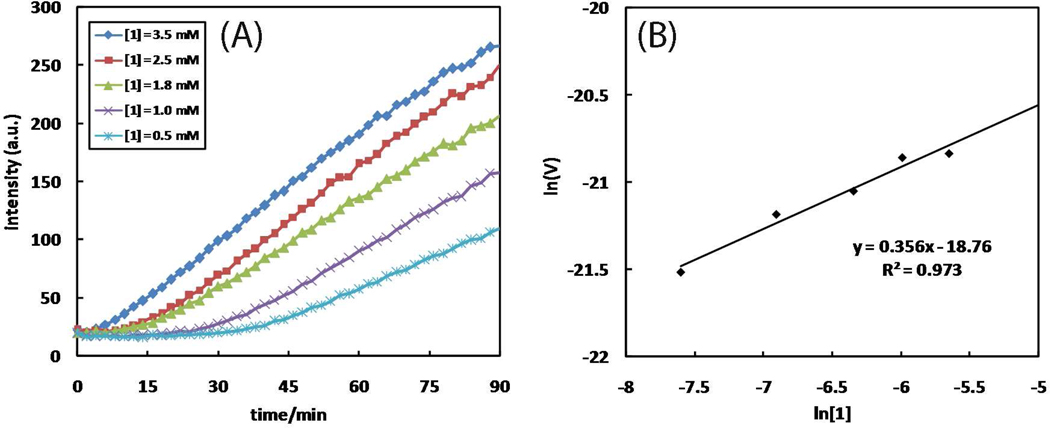
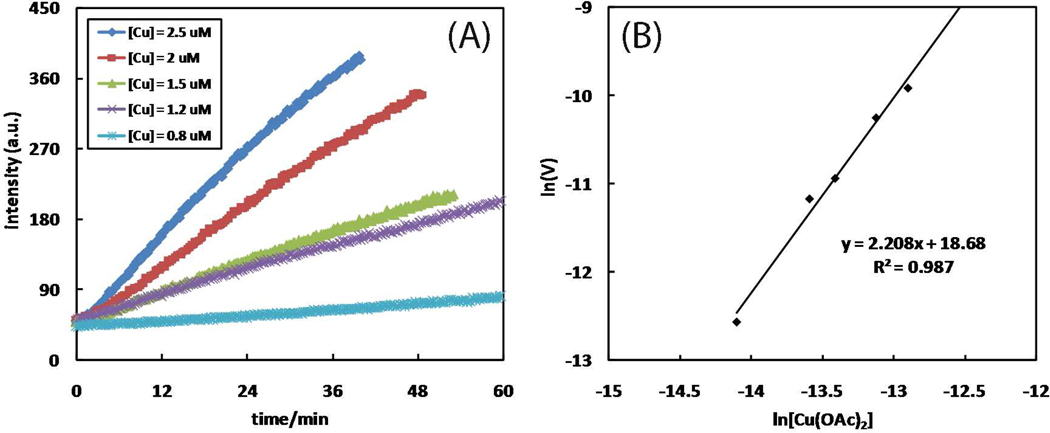
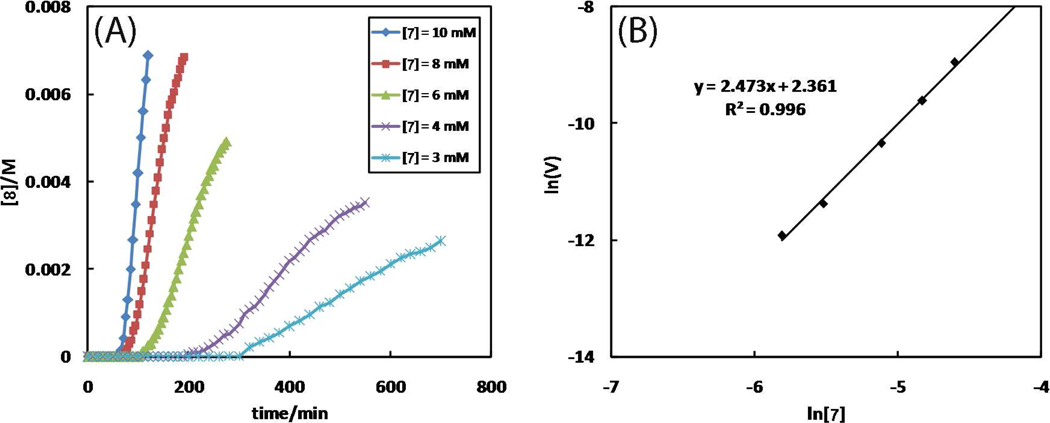
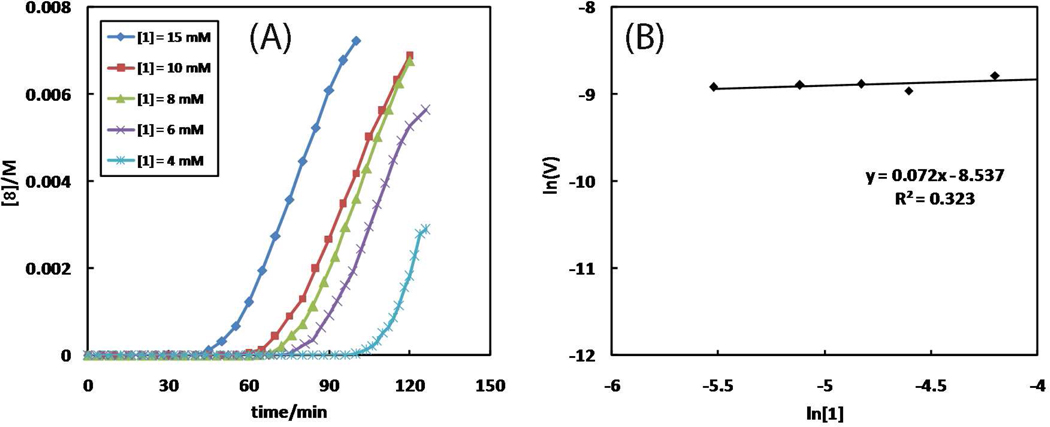
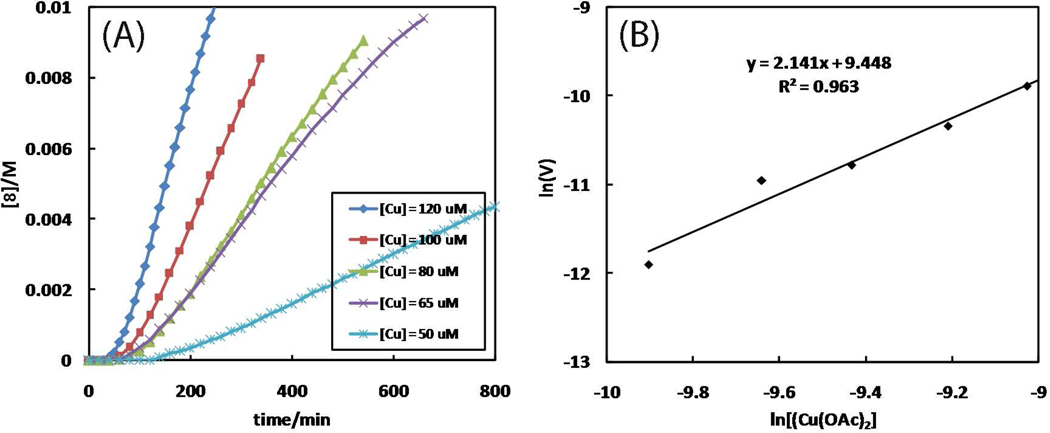
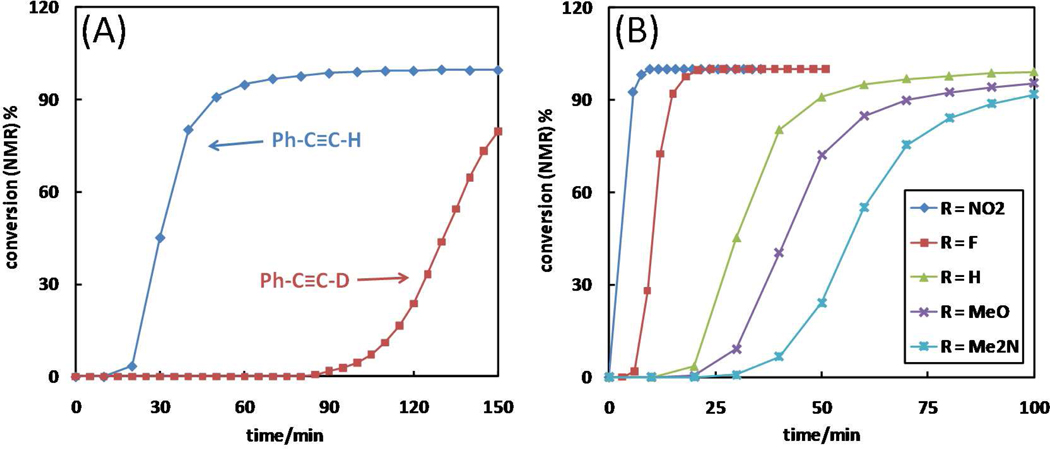
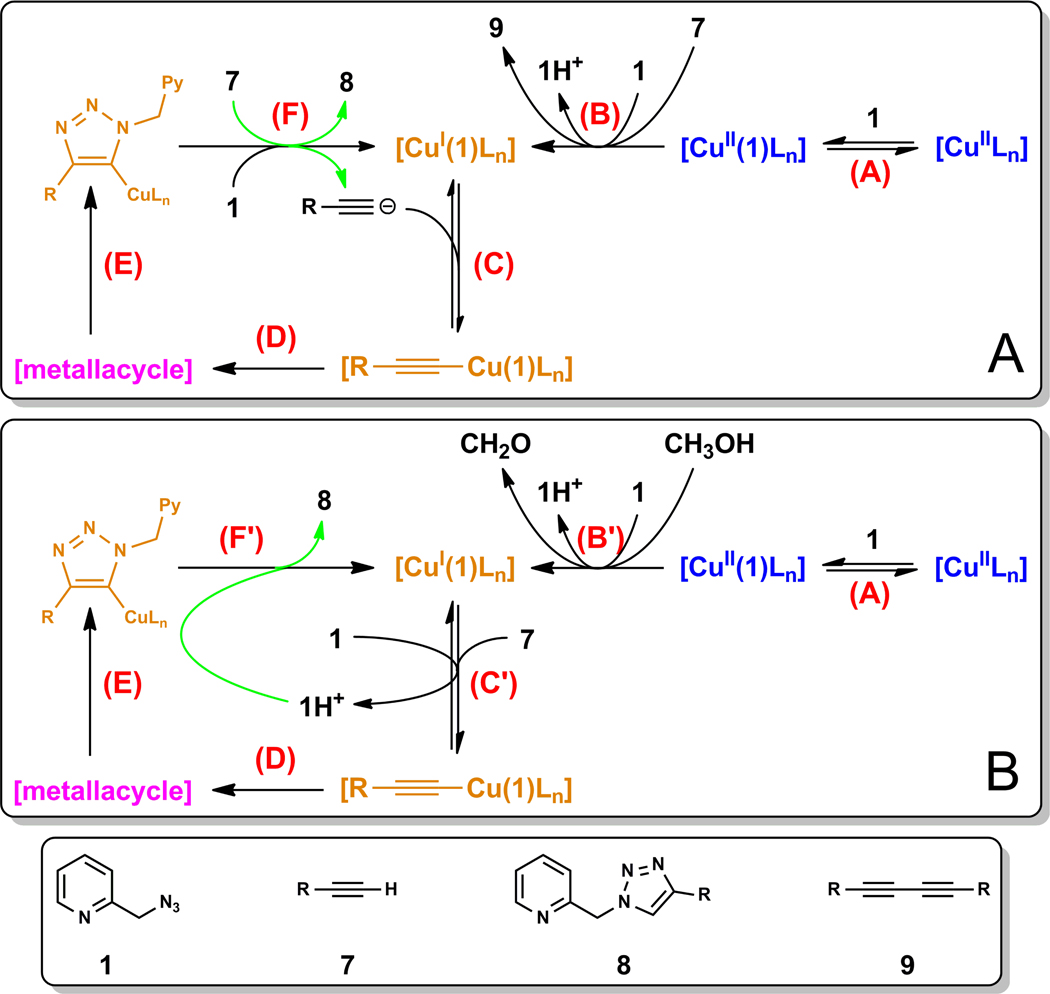
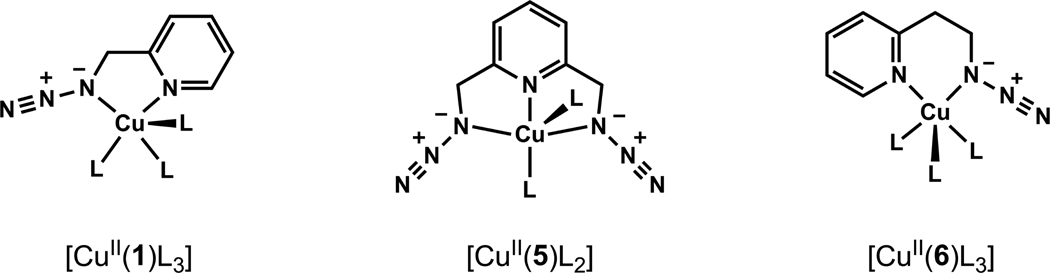
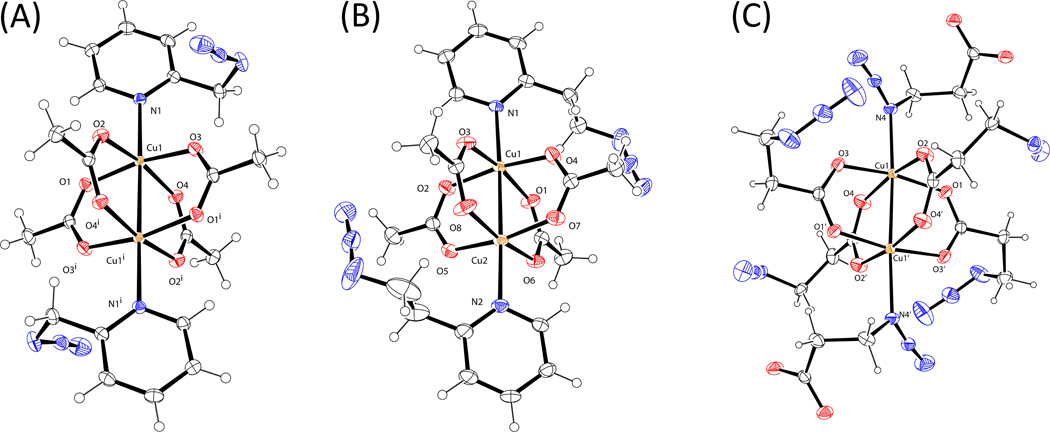
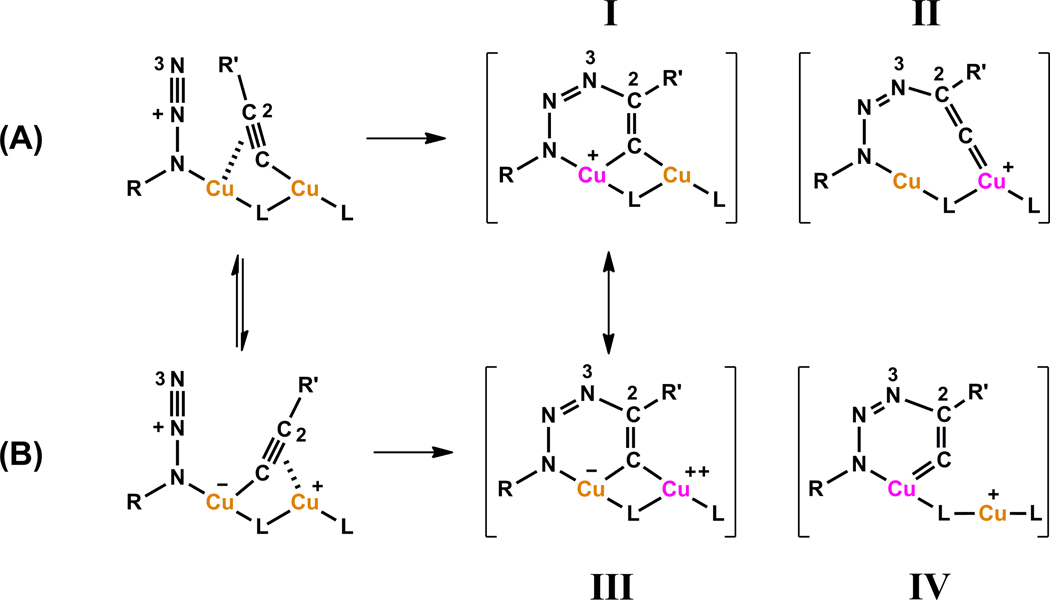
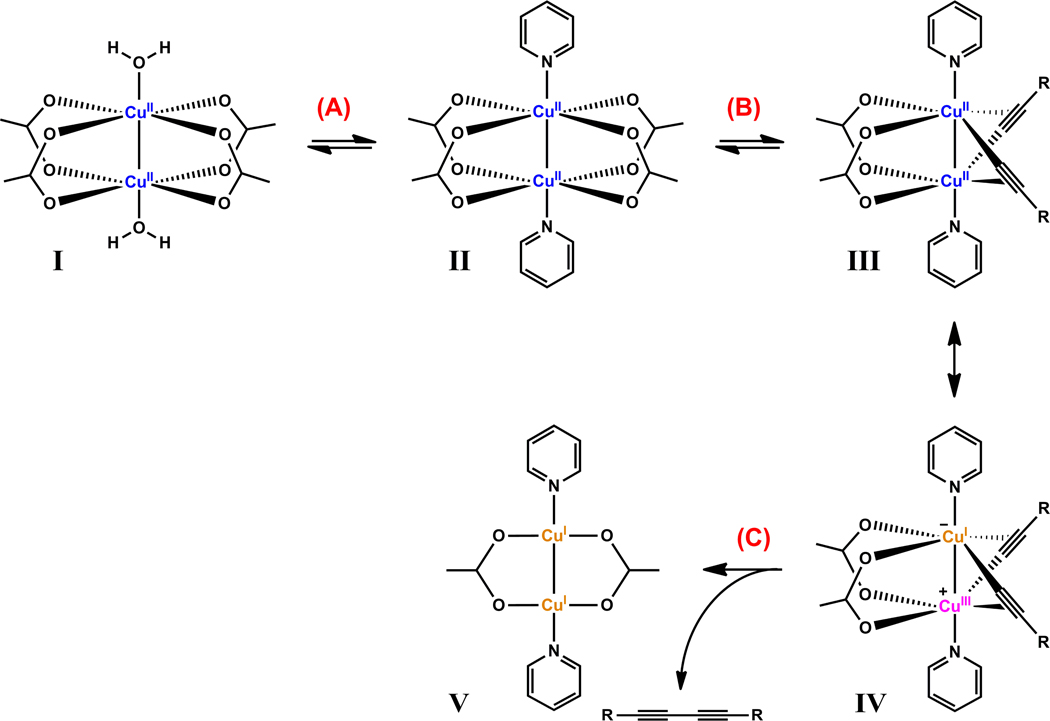
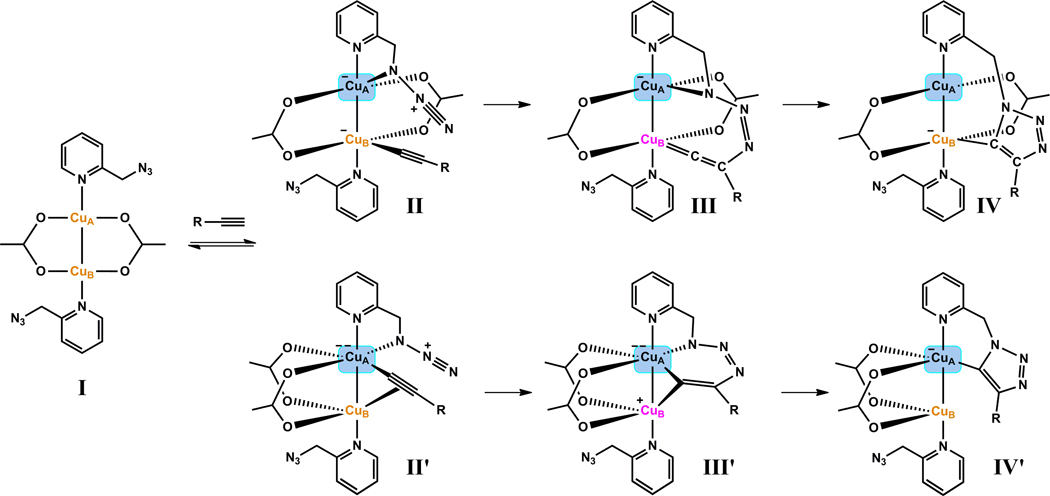
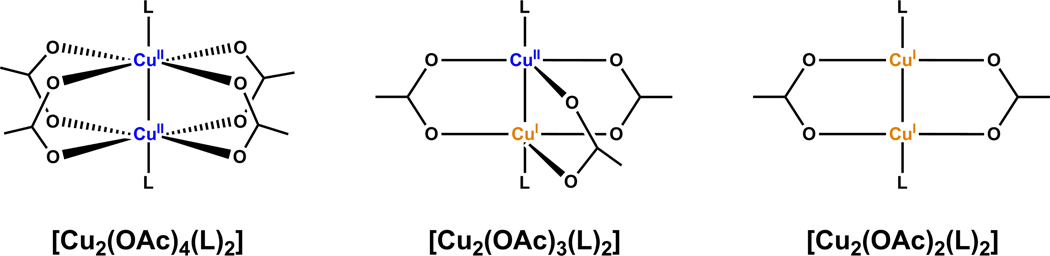
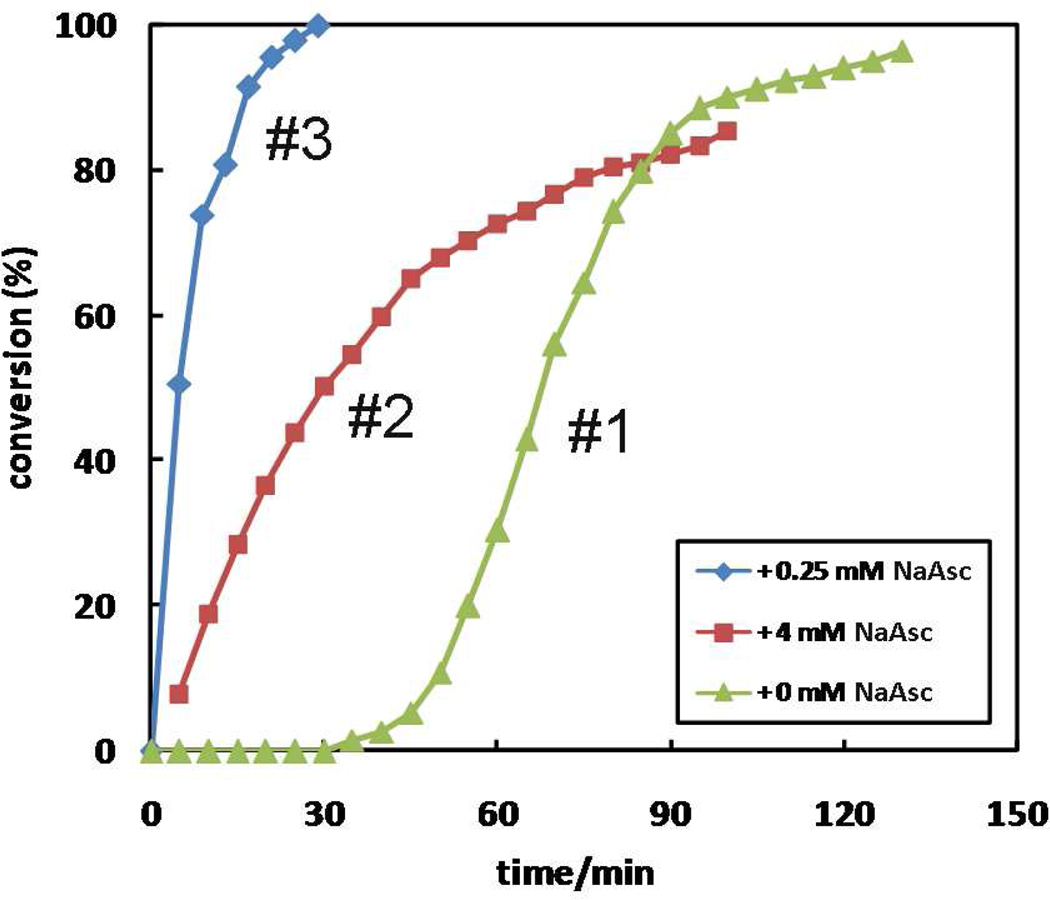
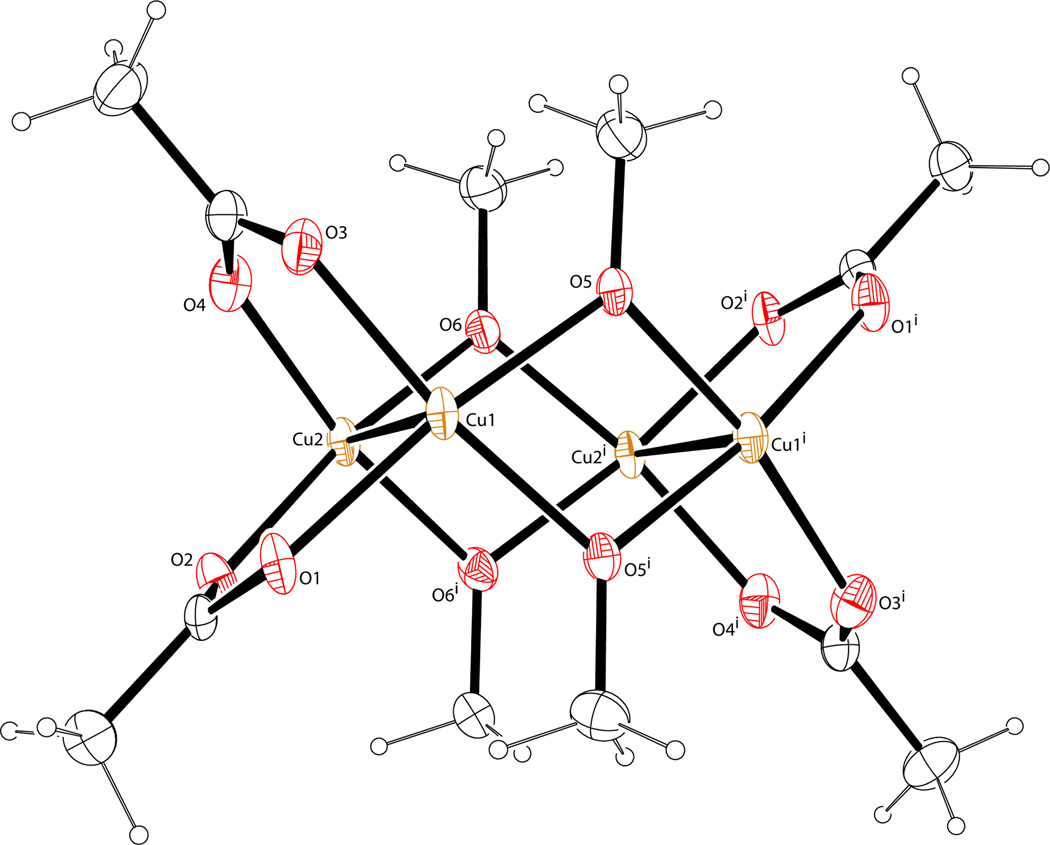
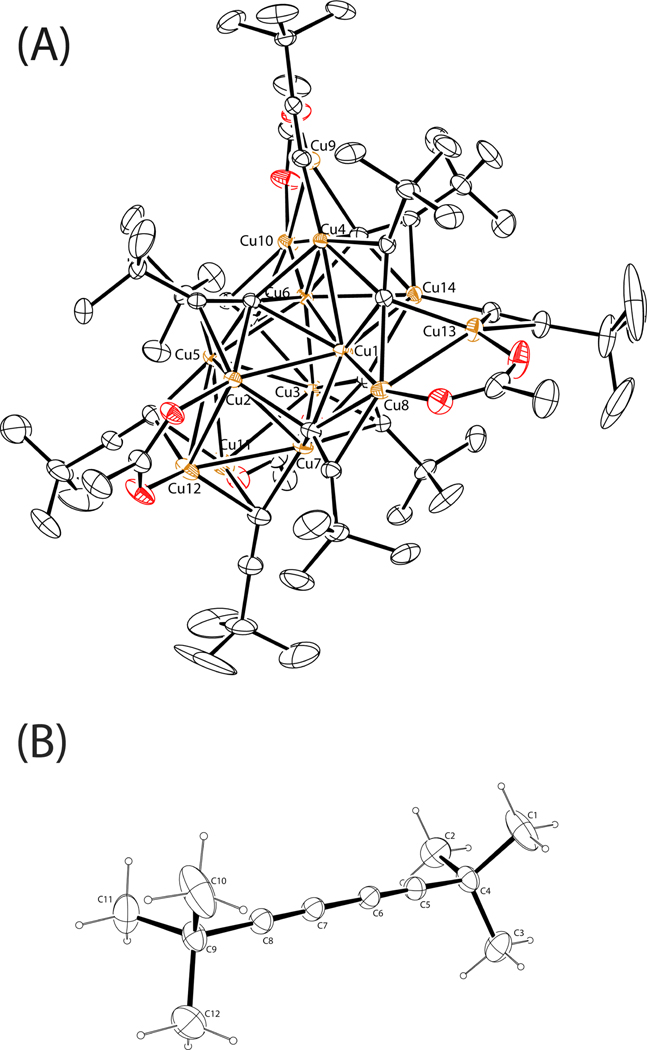
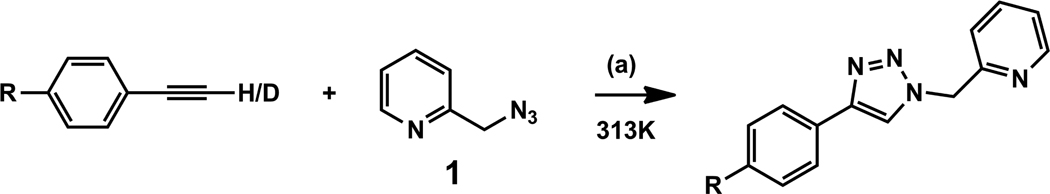
A mechanistic model is formulated to account for the high reactivity of chelating azides (organic azides capable of chelation-assisted metal coordination at the alkylated azido nitrogen position) and copper(II) acetate (Cu(OAc)(2)) in copper(II)-mediated azide-alkyne cycloaddition (AAC) reactions. Fluorescence and (1)H NMR assays are developed for monitoring the reaction progress in two different solvents, methanol and acetonitrile. Solvent kinetic isotopic effect and premixing experiments give credence to the proposed different induction reactions for converting copper(II) to catalytic copper(I) species in methanol (methanol oxidation) and acetonitrile (alkyne oxidative homocoupling), respectively. The kinetic orders of individual components in a chelation-assisted, copper(II)-accelerated AAC reaction are determined in both methanol and acetonitrile. Key conclusions resulting from the kinetic studies include (1) the interaction between copper ion (either in +1 or +2 oxidation state) and a chelating azide occurs in a fast, pre-equilibrium step prior to the formation of the in-cycle copper(I)-acetylide, (2) alkyne deprotonation is involved in several kinetically significant steps, and (3) consistent with prior experimental and computational results by other groups, two copper centers are involved in the catalysis. The X-ray crystal structures of chelating azides with Cu(OAc)(2) suggest a mechanistic synergy between alkyne oxidative homocoupling and copper(II)-accelerated AAC reactions, in which both a bimetallic catalytic pathway and a base are involved. The different roles of the two copper centers (a Lewis acid to enhance the electrophilicity of the azido group and a two-electron reducing agent in oxidative metallacycle formation, respectively) in the proposed catalytic cycle suggest that a mixed valency (+2 and +1) dinuclear copper species be a highly efficient catalyst. This proposition is supported by the higher activity of the partially reduced Cu(OAc)(2) in mediating a 2-picolylazide-involved AAC reaction than the fully reduced Cu(OAc)(2). Finally, the discontinuous kinetic behavior that has been observed by us and others in copper(I/II)-mediated AAC reactions is explained by the likely catalyst disintegration during the course of a relatively slow reaction. Complementing the prior mechanistic conclusions drawn by other investigators, which primarily focus on the copper(I)/alkyne interactions, we emphasize the kinetic significance of copper(I/II)/azide interaction. This work not only provides a mechanism accounting for the fast Cu(OAc)(2)-mediated AAC reactions involving chelating azides, which has apparent practical implications, but suggests the significance of mixed-valency dinuclear copper species in catalytic reactions where two copper centers carry different functions.
Figures
Similar articles
-
Chelation-assisted, copper(II)-acetate-accelerated azide-alkyne cycloaddition.J Org Chem. 2010 Oct 1;75(19):6540-8. doi: 10.1021/jo101305m. J Org Chem. 2010. PMID: 20806948 Free PMC article.
-
Apparent copper(II)-accelerated azide-alkyne cycloaddition.Org Lett. 2009 Nov 5;11(21):4954-7. doi: 10.1021/ol9021113. Org Lett. 2009. PMID: 19810690
-
Ligand-assisted, copper(II) acetate-accelerated azide-alkyne cycloaddition.Chem Asian J. 2011 Oct 4;6(10):2825-34. doi: 10.1002/asia.201100426. Epub 2011 Aug 29. Chem Asian J. 2011. PMID: 21954078
-
Development and Applications of the Copper-Catalyzed Azide-Alkyne Cycloaddition (CuAAC) as a Bioorthogonal Reaction.Molecules. 2016 Oct 24;21(10):1393. doi: 10.3390/molecules21101393. Molecules. 2016. PMID: 27783053 Free PMC article. Review.
-
Copper-catalyzed azide-alkyne cycloaddition (CuAAC) and beyond: new reactivity of copper(I) acetylides.Chem Soc Rev. 2010 Apr;39(4):1302-15. doi: 10.1039/b904091a. Epub 2010 Mar 4. Chem Soc Rev. 2010. PMID: 20309487 Free PMC article. Review.
Cited by
-
Bioorthogonal chemistry: strategies and recent developments.Chem Commun (Camb). 2013 Dec 7;49(94):11007-22. doi: 10.1039/c3cc44272a. Chem Commun (Camb). 2013. PMID: 24145483 Free PMC article. Review.
-
Single enantiomer propeller-shaped polynuclear complexes as catalysts-proof-of-concept for enantioinduction in a Michael addition reaction.R Soc Open Sci. 2025 Mar 27;12(3):241537. doi: 10.1098/rsos.241537. eCollection 2025 Mar. R Soc Open Sci. 2025. PMID: 40151485 Free PMC article.
-
On the regioselectivity of the mononuclear copper-catalyzed cycloaddition of azide and alkynes (CuAAC). A quantum chemical topological study.J Mol Model. 2014 Apr;20(4):2187. doi: 10.1007/s00894-014-2187-7. Epub 2014 Mar 25. J Mol Model. 2014. PMID: 24664121
-
Functionalized in Triplicate: A Ring-By-Ring Approach to Tailored Prodiginine Derivatives for Site-Specific Conjugation Through Click Chemistry.Chemistry. 2025 Aug 1;31(43):e202502066. doi: 10.1002/chem.202502066. Epub 2025 Jul 14. Chemistry. 2025. PMID: 40626904 Free PMC article.
-
New 1,2,3-Triazoles from (R)-Carvone: Synthesis, DFT Mechanistic Study and In Vitro Cytotoxic Evaluation.Molecules. 2022 Jan 25;27(3):769. doi: 10.3390/molecules27030769. Molecules. 2022. PMID: 35164037 Free PMC article.
References
-
- Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew. Chem. Int. Ed. 2002;41:2596–2599. - PubMed
-
- Tornøe CW, Christensen C, Meldal M. J. Org. Chem. 2002;67:3057–3064. - PubMed
-
- Wu P, Fokin VV. Aldrichimica Acta. 2007;40:7–17.
-
- Moses JE, Moorhouse AD. Chem. Soc. Rev. 2007;36:1249–1262. - PubMed
-
- Meldal M, Tornøe CW. Chem. Rev. 2008;108:2952–3015. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
