

Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes
- PMID: 21810661
- PMCID: PMC3167235
- DOI: 10.1161/CIRCULATIONAHA.110.005405
Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes
Abstract
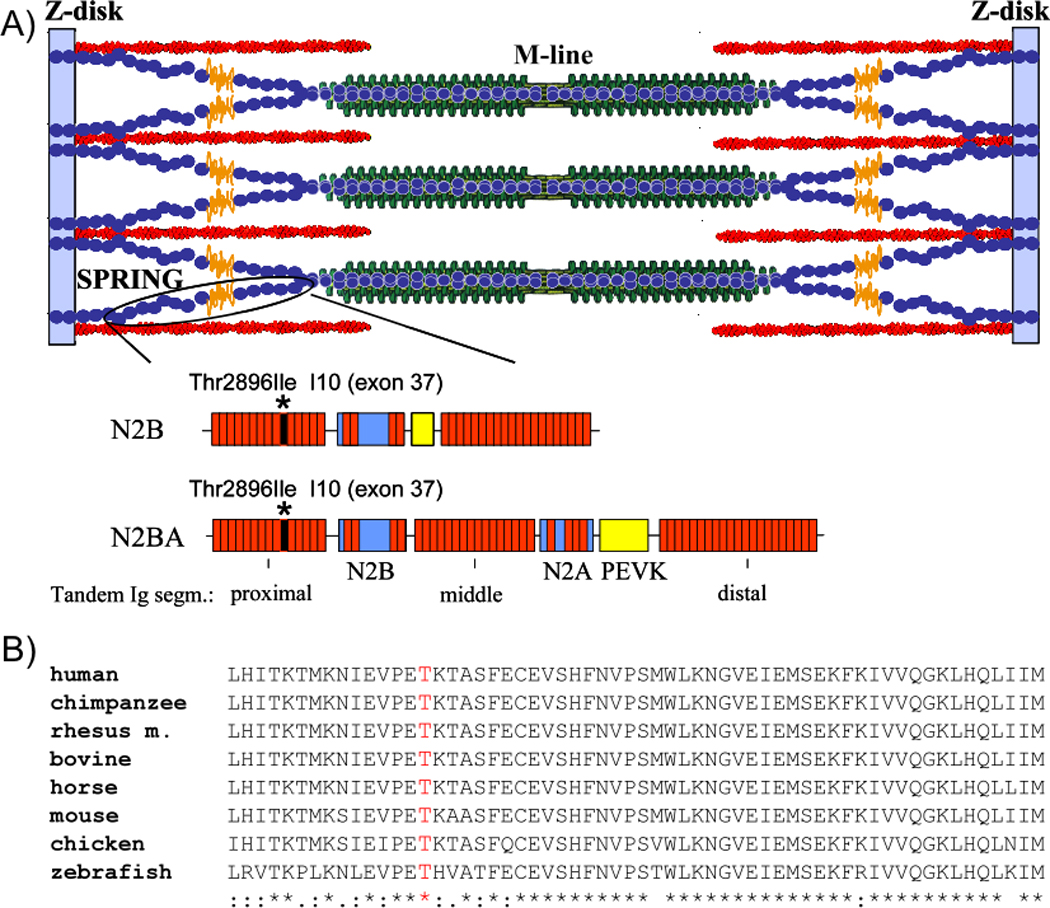
Background: Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited genetic myocardial disease characterized by fibrofatty replacement of the myocardium and a predisposition to cardiac arrhythmias and sudden death. We evaluated the cardiomyopathy gene titin (TTN) as a candidate ARVC gene because of its proximity to an ARVC locus at position 2q32 and the connection of the titin protein to the transitional junction at intercalated disks.
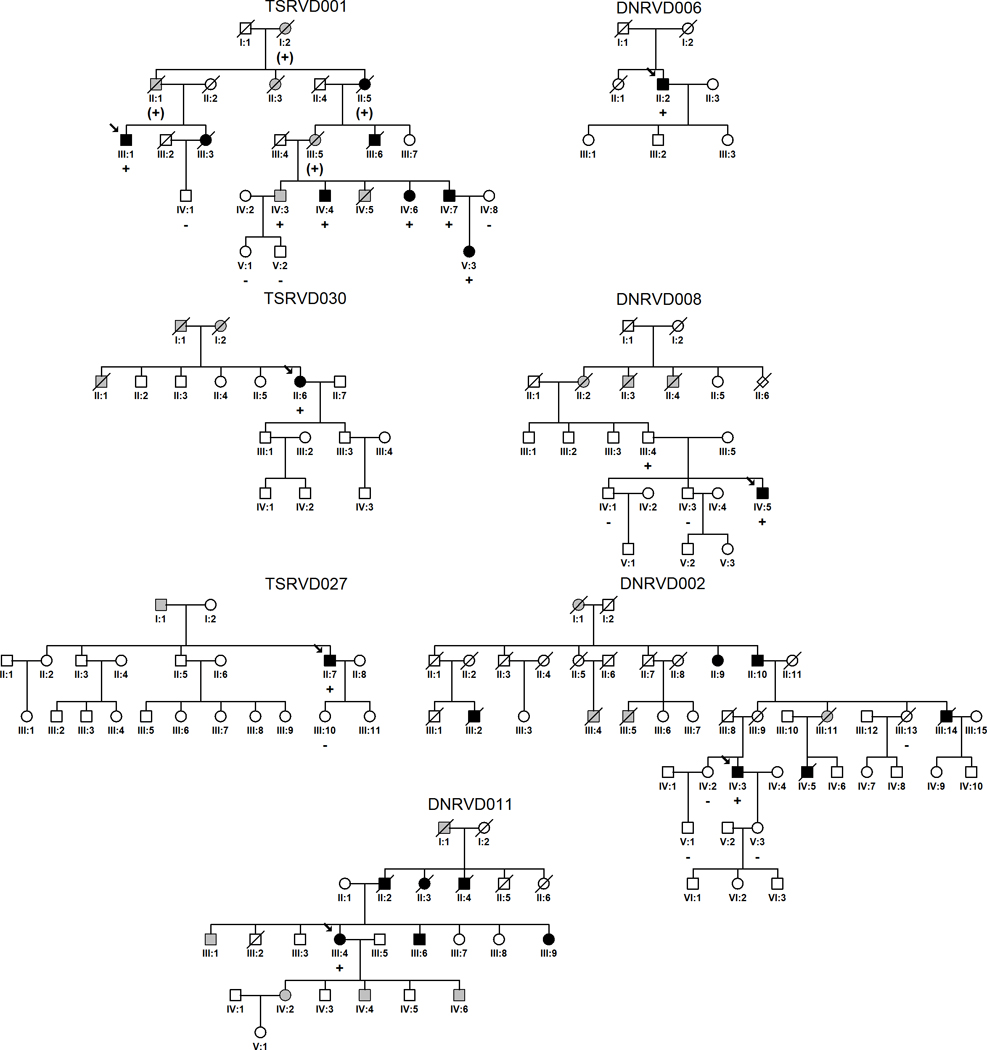
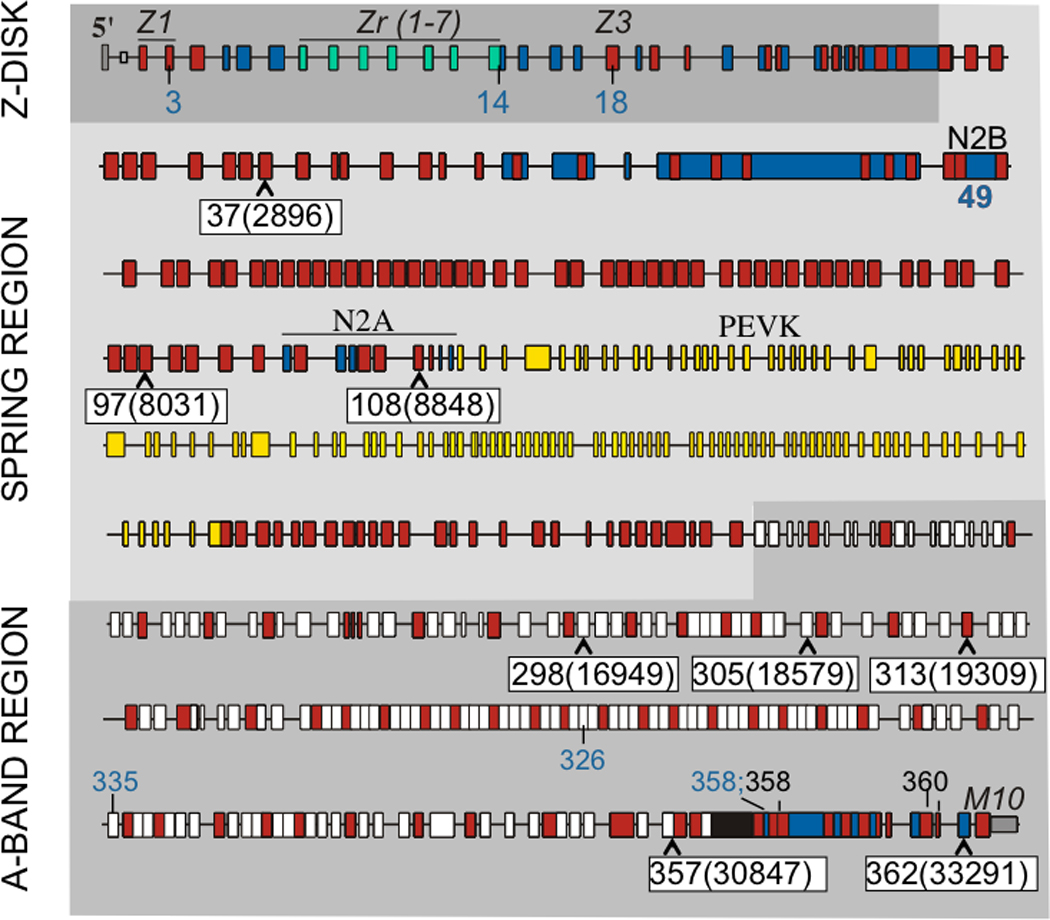
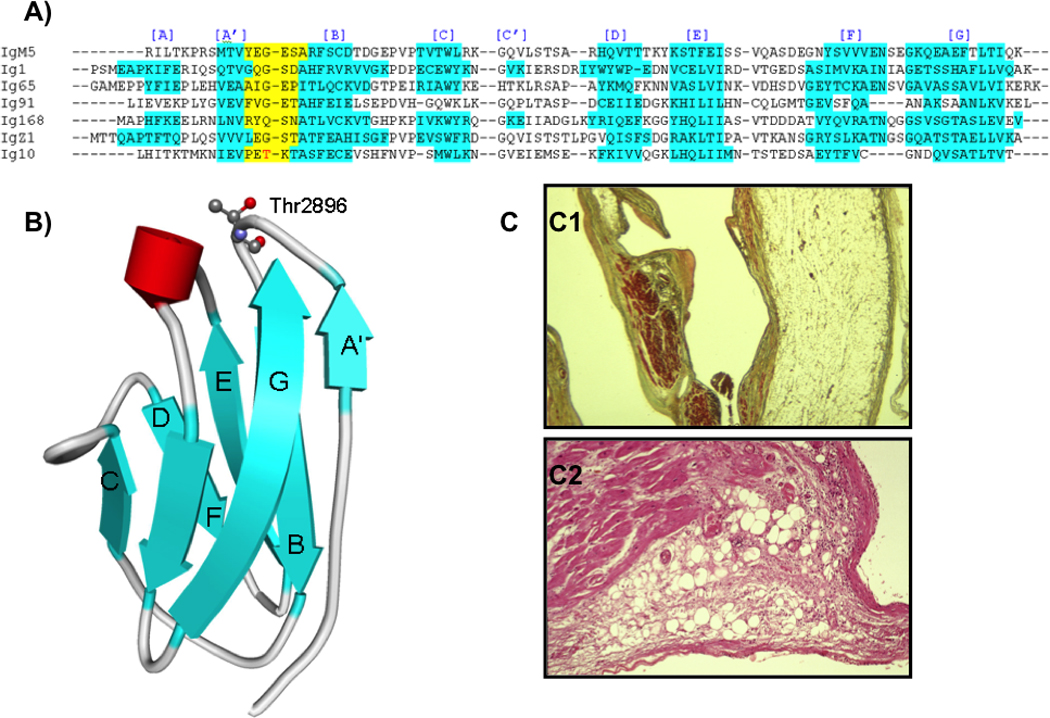
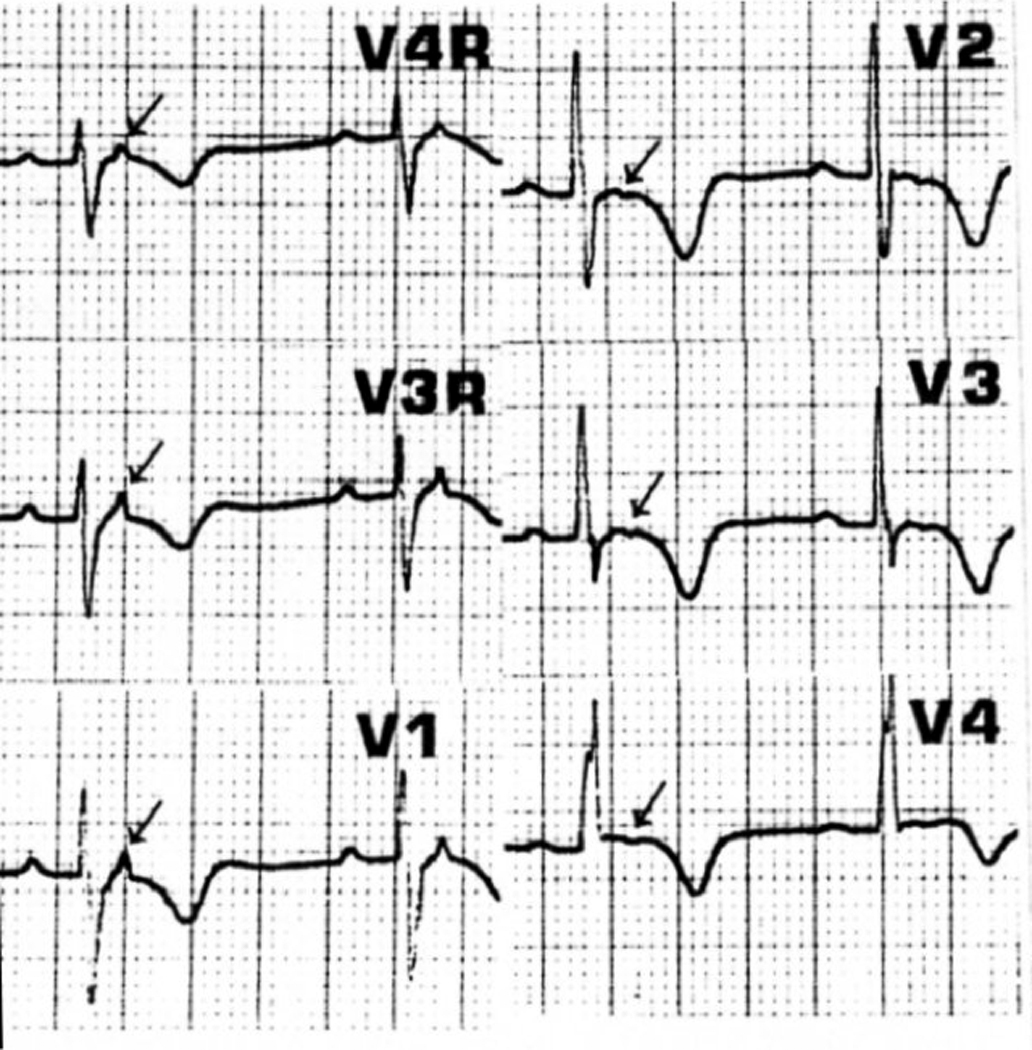
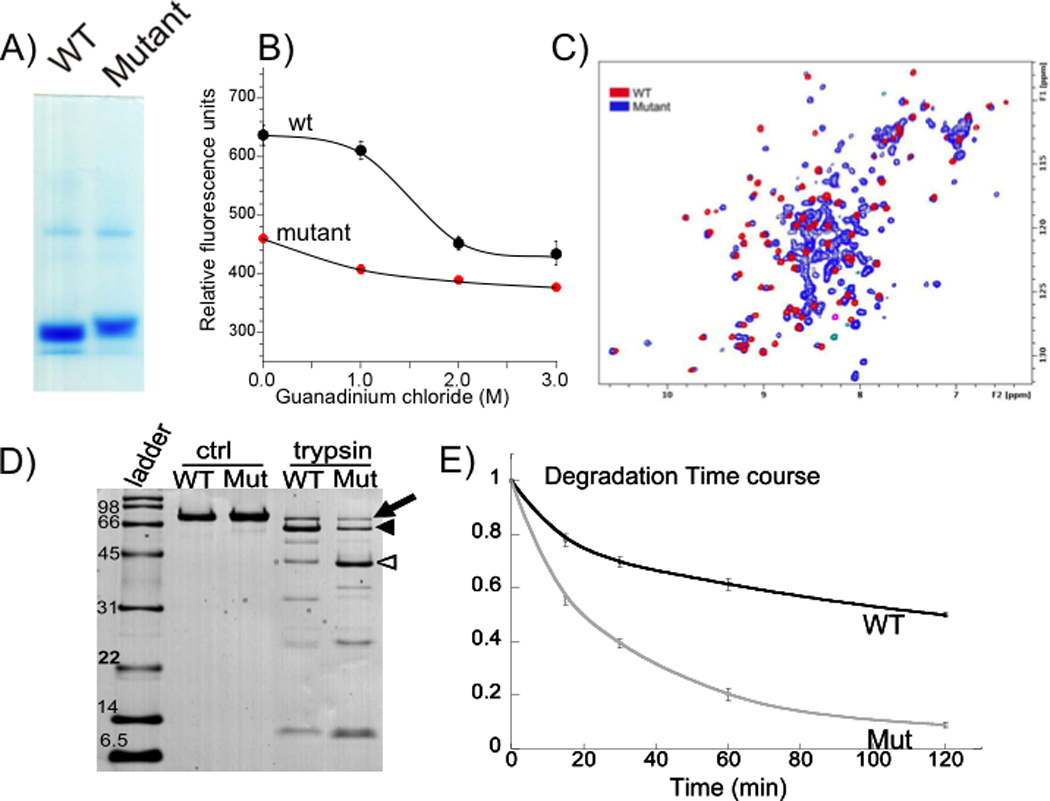
Methods and results: All 312 titin exons known to be expressed in human cardiac titin and the complete 3' untranslated region were sequenced in 38 ARVC families. Eight unique TTN variants were detected in 7 families, including a prominent Thr2896Ile mutation that showed complete segregation with the ARVC phenotype in 1 large family. The Thr2896IIe mutation maps within a highly conserved immunoglobulin-like fold (Ig10 domain) located in the spring region of titin. Native gel electrophoresis, nuclear magnetic resonance, intrinsic fluorescence, and proteolysis assays of wild-type and mutant Ig10 domains revealed that the Thr2896IIe exchange reduces the structural stability and increases the propensity for degradation of the Ig10 domain. The phenotype of TTN variant carriers was characterized by a history of sudden death (5 of 7 families), progressive myocardial dysfunction causing death or heart transplantation (8 of 14 cases), frequent conduction disease (11 of 14), and incomplete penetrance (86%).
Conclusions: Our data provide evidence that titin mutations can cause ARVC, a finding that further expands the origin of the disease beyond desmosomal proteins. Structural impairment of the titin spring is a likely cause of ARVC and constitutes a novel mechanism underlying myocardial remodeling and sudden cardiac death.
Figures
References
-
- Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. - PubMed
-
- Nava A, Thiene G, Canciani B, Scognamiglio R, Daliento L, Buja G, Maertini B, Stritoni P, Fasoli G. Familial occurrence of right ventricular dysplasia. A study involving nine families. J Am Coll Cardiol. 1988;12:1222–1228. - PubMed
-
- den Haan AD, Tan BY, Zikusoka MN, Llado LI, Jain R, Daly A, Tichnell C, James C, Amat-Alarcon N, Abraham T, Russell SD, Bluemke DA, Calkins H, Dalal D, Judge DP. Comprehensive desmosome mutation analysis in north americans with arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Cardiovasc Genet. 2009;2:428–435. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
