

Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation
- PMID: 21811164
- PMCID: PMC3398737
- DOI: 10.1097/GIM.0b013e318226fbf2
Targeted polymerase chain reaction-based enrichment and next generation sequencing for diagnostic testing of congenital disorders of glycosylation
Abstract
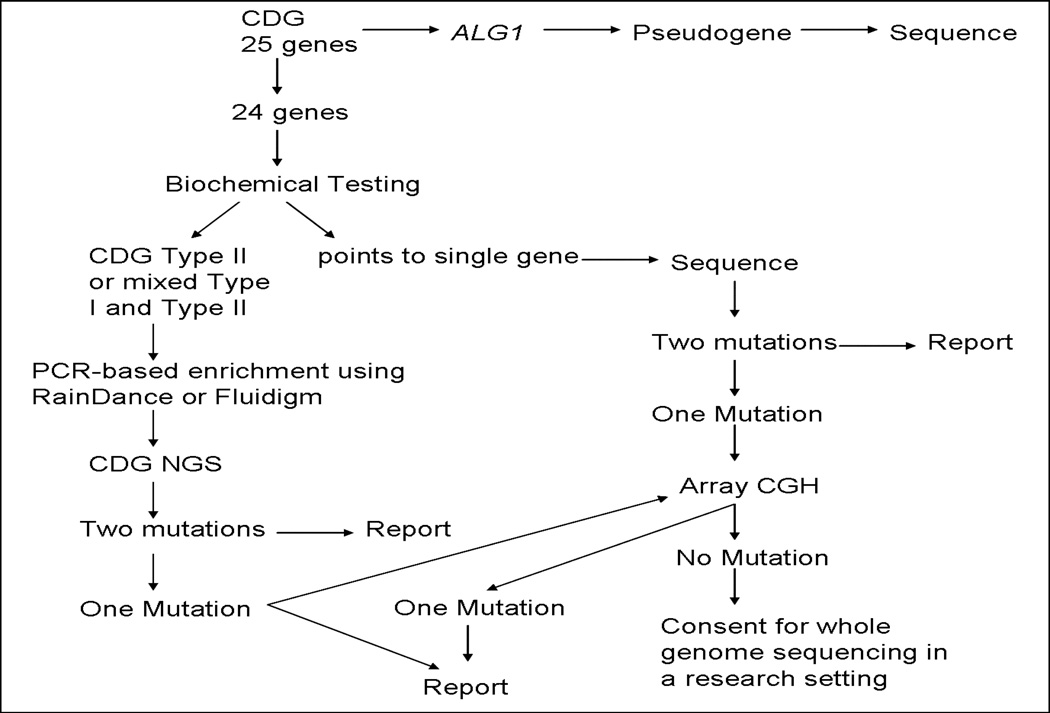
Purpose: Congenital disorders of glycosylation are a heterogeneous group of disorders caused by deficient glycosylation, primarily affecting the N-linked pathway. It is estimated that more than 40% of congenital disorders of glycosylation patients lack a confirmatory molecular diagnosis. The purpose of this study was to improve molecular diagnosis for congenital disorders of glycosylation by developing and validating a next generation sequencing panel for comprehensive mutation detection in 24 genes known to cause congenital disorders of glycosylation.
Methods: Next generation sequencing validation was performed on 12 positive control congenital disorders of glycosylation patients. These samples were blinded as to the disease-causing mutations. Both RainDance and Fluidigm platforms were used for sequence enrichment and targeted amplification. The SOLiD platform was used for sequencing the amplified products. Bioinformatic analysis was performed using NextGENe® software.
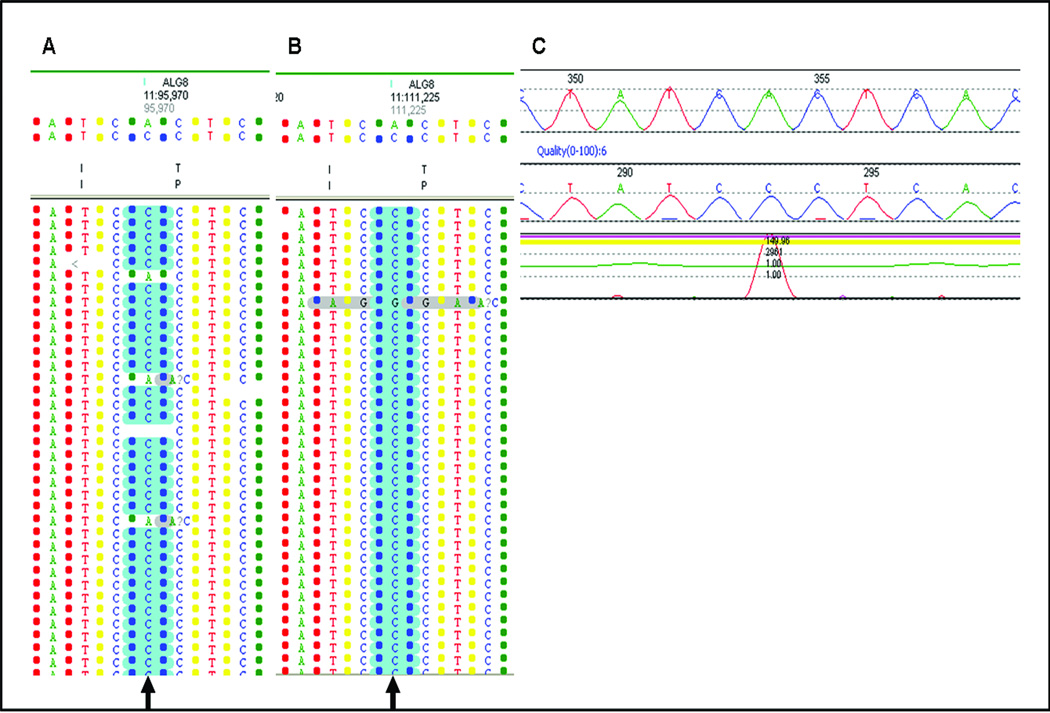
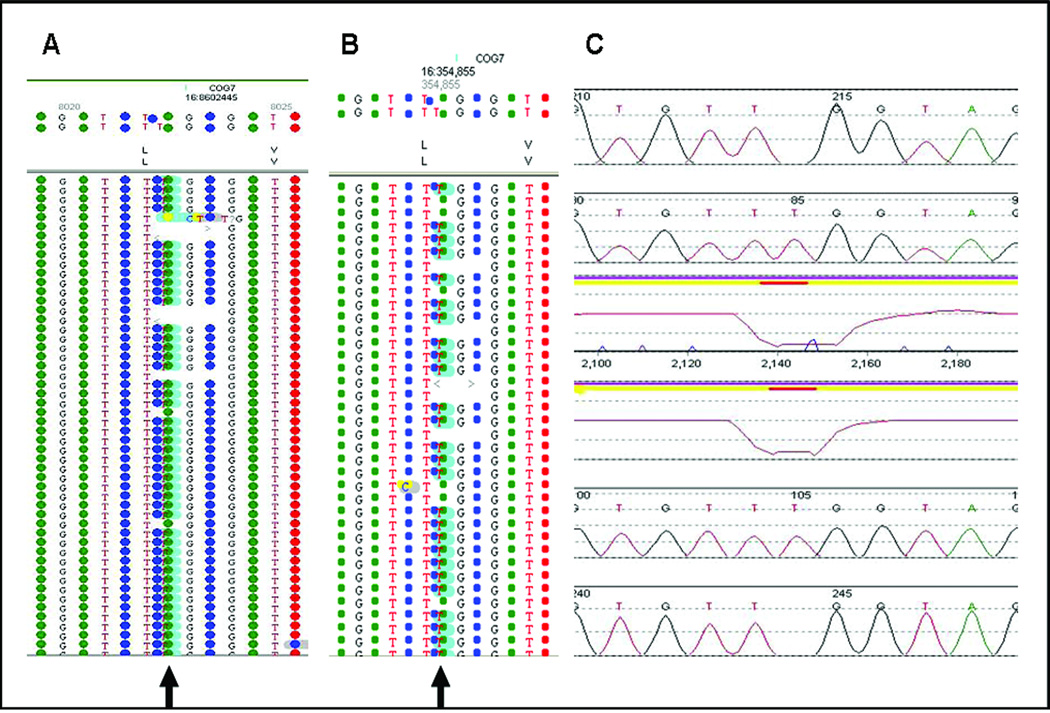
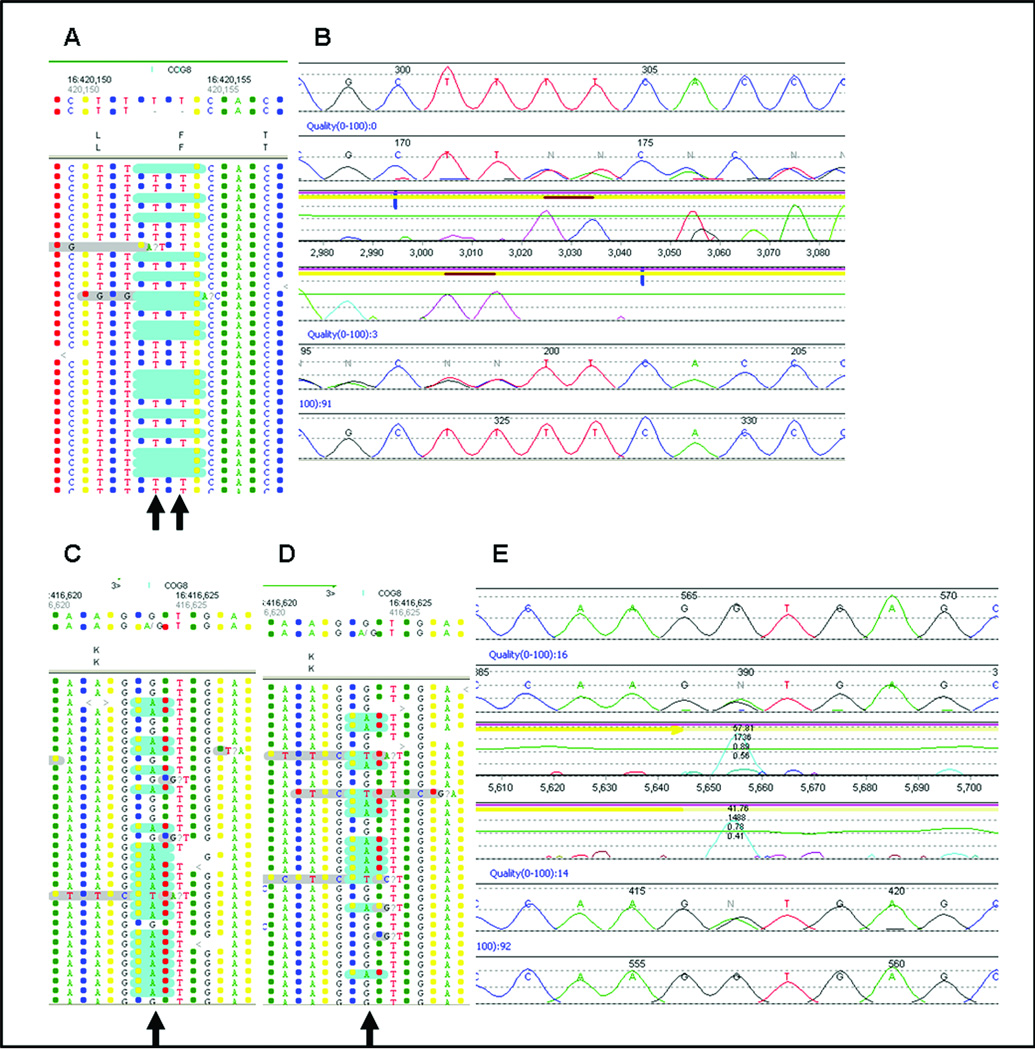
Results: The disease-causing mutations were identified by next generation sequencing for all 12 positive controls. Additional variants were also detected in three controls that are known or predicted to impair gene function and may contribute to the clinical phenotype.
Conclusions: We conclude that development of next generation sequencing panels in the diagnostic laboratory where multiple genes are implicated in a disorder is more cost-effective and will result in improved and faster patient diagnosis compared with a gene-by-gene approach. Recommendations are also provided for data analysis from the next generation sequencing-derived data in the clinical laboratory, which will be important for the widespread use of this technology.
Conflict of interest statement
The authors declare no conflicts of interest
Figures
References
-
- Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999 Dec 6;1473(1):4–8. - PubMed
-
- Freeze HH. Genetic defects in the human glycome. Nat Rev Genet. 2006 Jul;7(7):537–551. - PubMed
-
- Jaeken J, Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu Rev Genomics Hum Genet. 2007;8:261–278. - PubMed
-
- Footitt EJ, Karimova A, Burch M, et al. Cardiomyopathy in the congenital disorders of glycosylation (CDG): a case of late presentation and literature review. J Inherit Metab Dis. 2009 Sep 7; - PubMed
Publication types
MeSH terms
Grants and funding
- KL2 TR000455/TR/NCATS NIH HHS/United States
- R01 DK055615/DK/NIDDK NIH HHS/United States
- TL1 RR025010/RR/NCRR NIH HHS/United States
- KL2 RR025009/RR/NCRR NIH HHS/United States
- MDA G6396330/PHS HHS/United States
- TL1 TR000456/TR/NCATS NIH HHS/United States
- UL1 TR000454/TR/NCATS NIH HHS/United States
- RC1 NS069541/NS/NINDS NIH HHS/United States
- KL2 R0025009/PHS HHS/United States
- T32 MH087977/MH/NIMH NIH HHS/United States
- UL1 RR025008/RR/NCRR NIH HHS/United States
- RC1NS069541-01/NS/NINDS NIH HHS/United States
- 1T32MH087977/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
