

Learning to predict chemical reactions
- PMID: 21819139
- PMCID: PMC3193800
- DOI: 10.1021/ci200207y
Learning to predict chemical reactions
Abstract
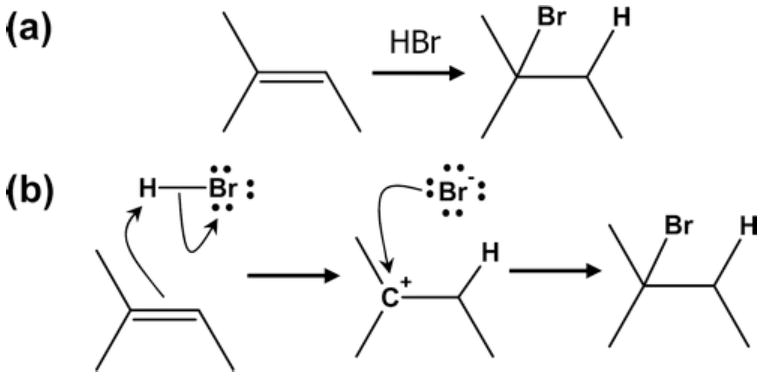
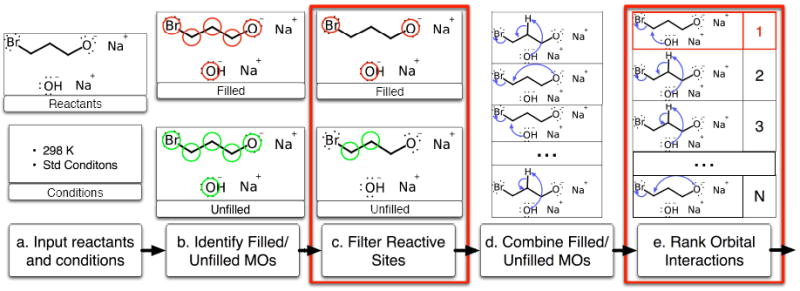
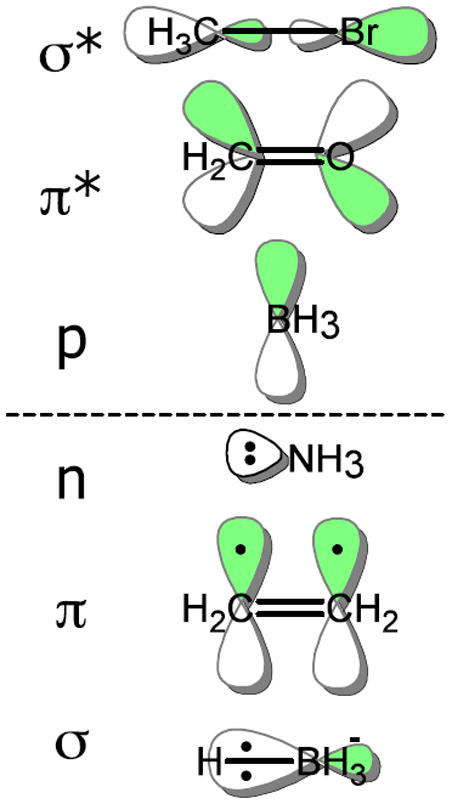
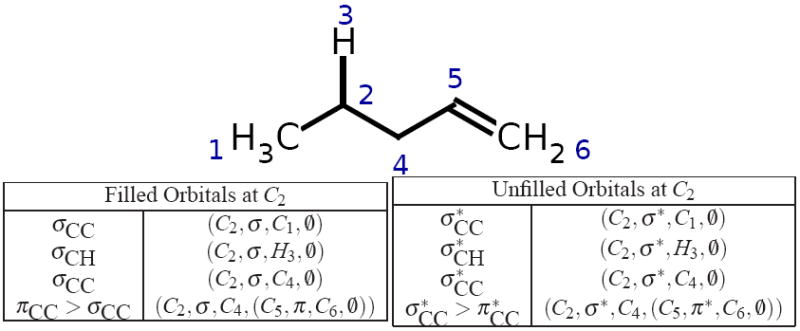
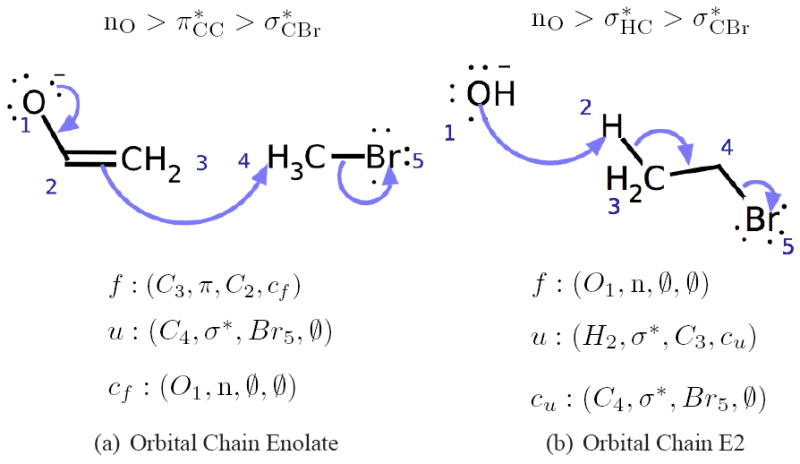
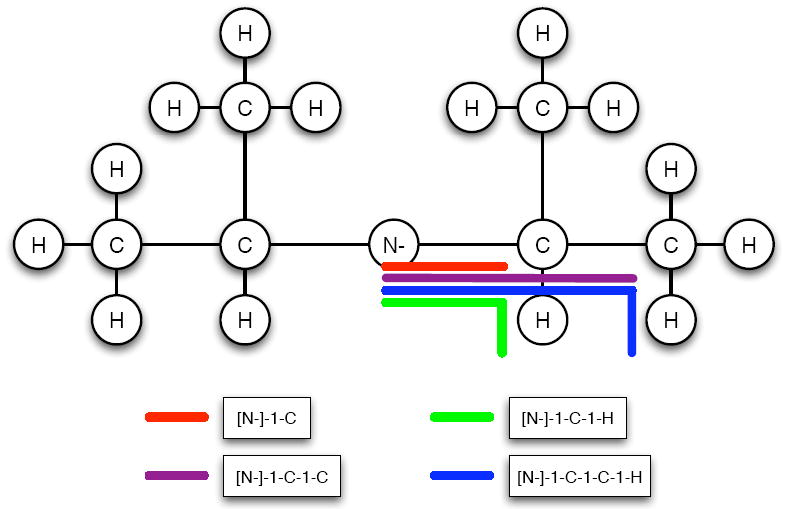
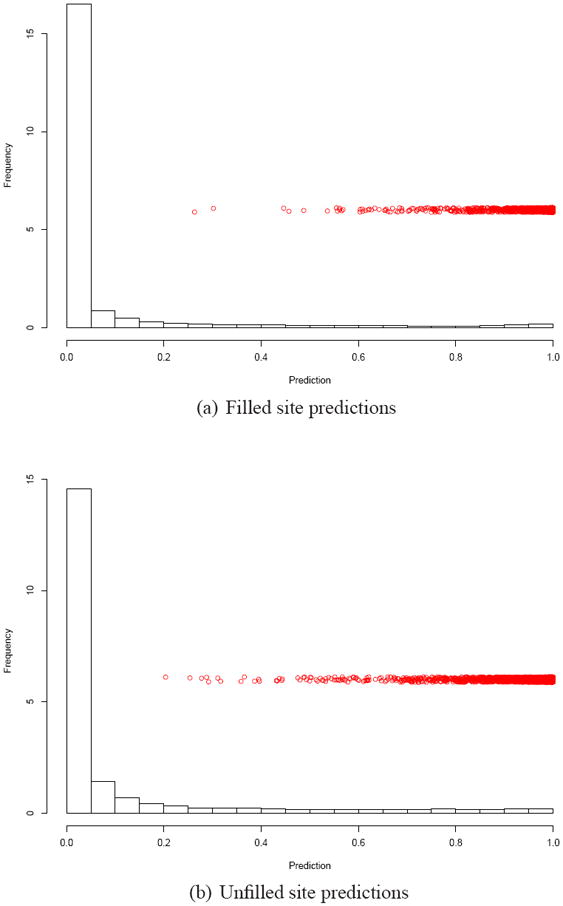
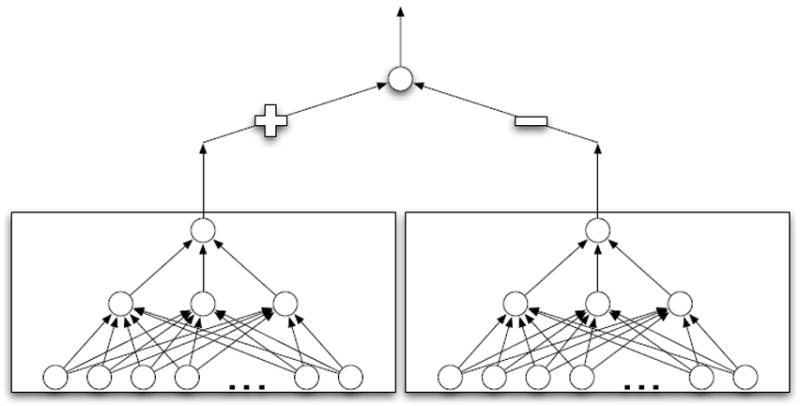
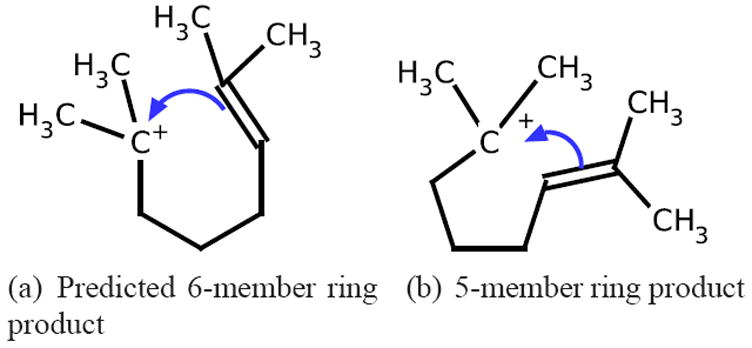
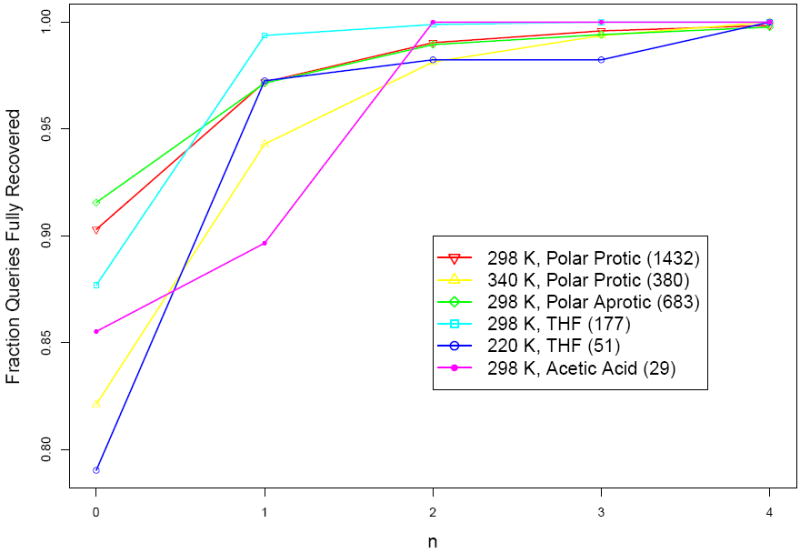
Being able to predict the course of arbitrary chemical reactions is essential to the theory and applications of organic chemistry. Approaches to the reaction prediction problems can be organized around three poles corresponding to: (1) physical laws; (2) rule-based expert systems; and (3) inductive machine learning. Previous approaches at these poles, respectively, are not high throughput, are not generalizable or scalable, and lack sufficient data and structure to be implemented. We propose a new approach to reaction prediction utilizing elements from each pole. Using a physically inspired conceptualization, we describe single mechanistic reactions as interactions between coarse approximations of molecular orbitals (MOs) and use topological and physicochemical attributes as descriptors. Using an existing rule-based system (Reaction Explorer), we derive a restricted chemistry data set consisting of 1630 full multistep reactions with 2358 distinct starting materials and intermediates, associated with 2989 productive mechanistic steps and 6.14 million unproductive mechanistic steps. And from machine learning, we pose identifying productive mechanistic steps as a statistical ranking, information retrieval problem: given a set of reactants and a description of conditions, learn a ranking model over potential filled-to-unfilled MO interactions such that the top-ranked mechanistic steps yield the major products. The machine learning implementation follows a two-stage approach, in which we first train atom level reactivity filters to prune 94.00% of nonproductive reactions with a 0.01% error rate. Then, we train an ensemble of ranking models on pairs of interacting MOs to learn a relative productivity function over mechanistic steps in a given system. Without the use of explicit transformation patterns, the ensemble perfectly ranks the productive mechanism at the top 89.05% of the time, rising to 99.86% of the time when the top four are considered. Furthermore, the system is generalizable, making reasonable predictions over reactants and conditions which the rule-based expert does not handle. A web interface to the machine learning based mechanistic reaction predictor is accessible through our chemoinformatics portal ( http://cdb.ics.uci.edu) under the Toolkits section.
Figures
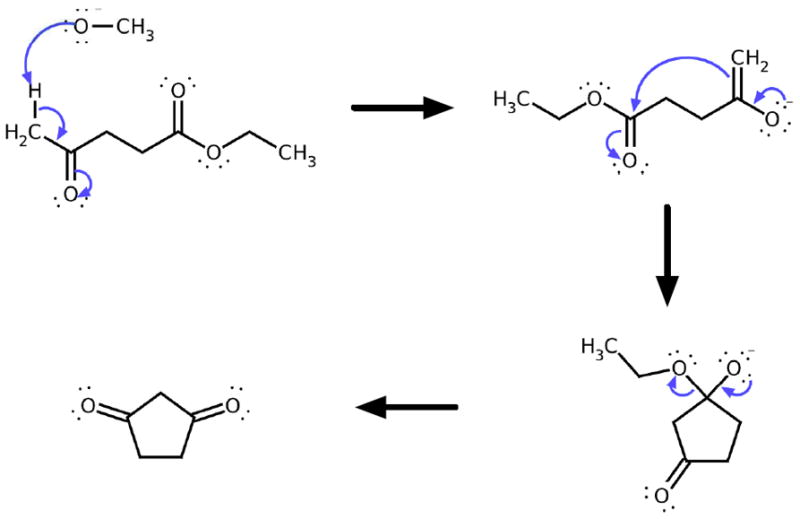
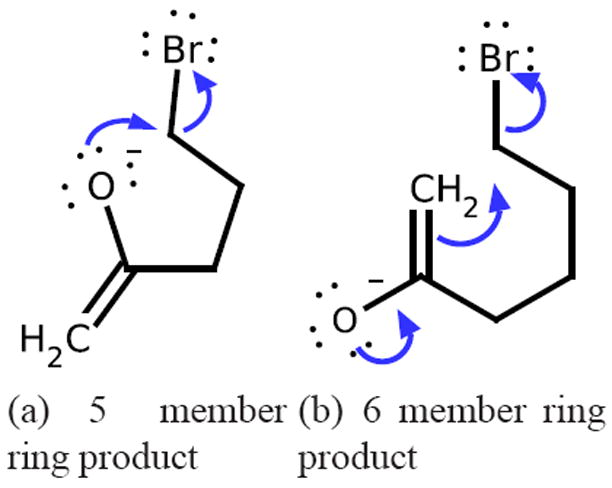
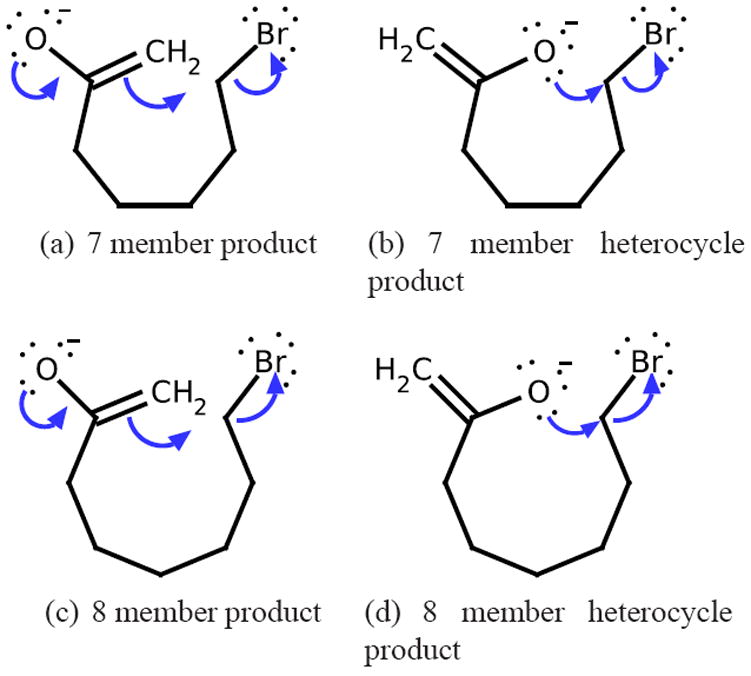
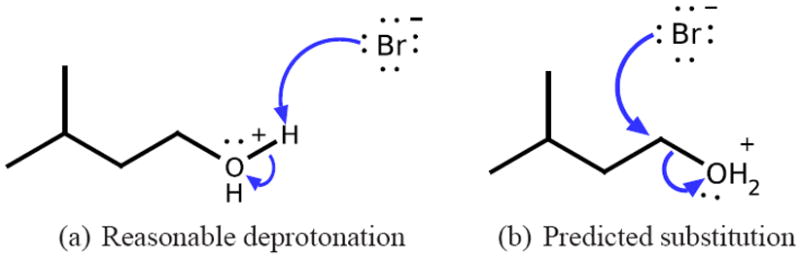
References
-
- Lu Z, Yang W. Reaction path potential for complex systems derived from combined ab initio quantum mechanical and molecular mechanical calculations. J Chem Phys. 2004;121:89–100. - PubMed
-
- Peters B, Heyden A, Bell AT, Chakraborty A. A growing string method for determining transition states: comparison to the nudged elastic band and string methods. J Chem Phys. 2004;120:7877–7786. - PubMed
-
- Cramer C. Essentials of Computational Chemistry: Theories and Models. 2. Wiley; West Sussex, England: 2004.
-
- Henkelman G, Uberuaga BP, Jónsson H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J Chem Phys. 2000;113:9901–9904.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
