

Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome
- PMID: 21820096
- PMCID: PMC3155174
- DOI: 10.1016/j.ajhg.2011.07.009
Recessive mutations in DOCK6, encoding the guanidine nucleotide exchange factor DOCK6, lead to abnormal actin cytoskeleton organization and Adams-Oliver syndrome
Abstract
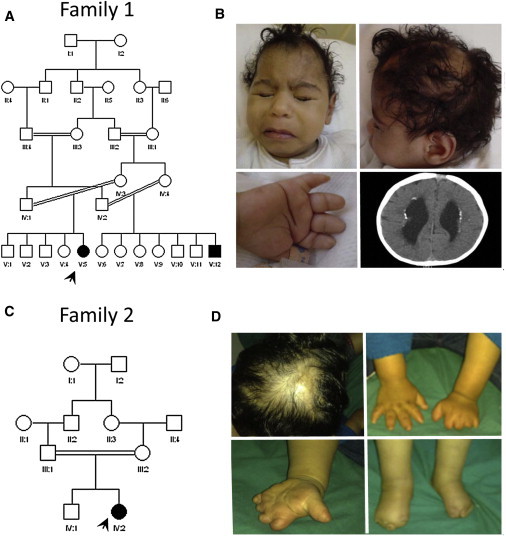
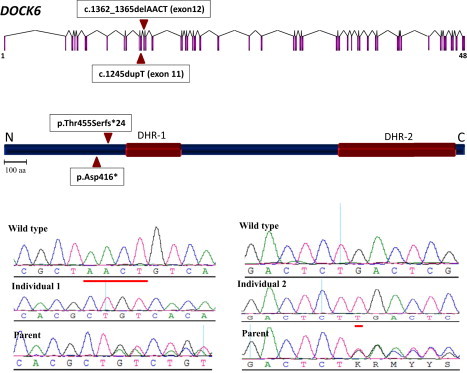
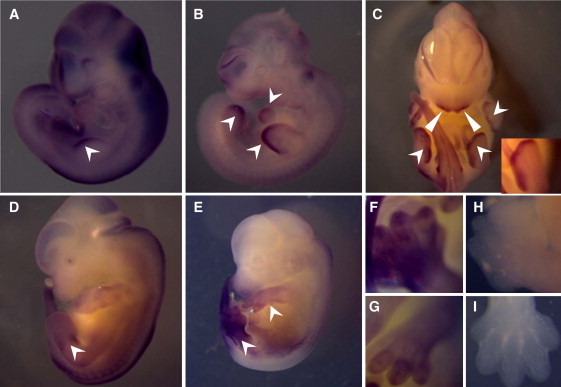
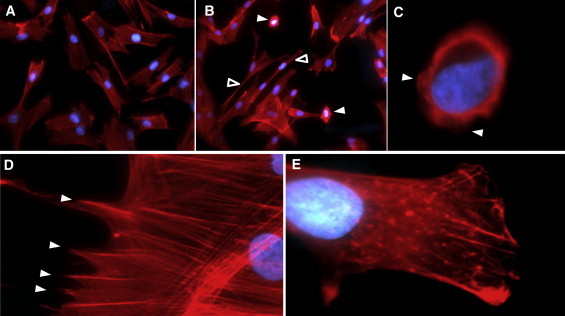
Adams-Oliver syndrome (AOS) is defined by the combination of aplasia cutis congenita (ACC) and terminal transverse limb defects (TTLD). It is usually inherited as an autosomal-dominant trait, but autosomal-recessive inheritance has also been documented. In an individual with autosomal-recessive AOS, we combined autozygome analysis with exome sequencing to identify a homozygous truncating mutation in dedicator of cytokinesis 6 gene (DOCK6) which encodes an atypical guanidine exchange factor (GEF) known to activate two members of the Rho GTPase family: Cdc42 and Rac1. Another homozygous truncating mutation was identified upon targeted sequencing of DOCK6 in an unrelated individual with AOS. Consistent with the established role of Cdc42 and Rac1 in the organization of the actin cytoskeleton, we demonstrate a cellular phenotype typical of a defective actin cytoskeleton in patient cells. These findings, combined with a Dock6 expression profile that is consistent with an AOS phenotype as well as the very recent demonstration of dominant mutations of ARHGAP31 in AOS, establish Cdc42 and Rac1 as key molecules in the pathogenesis of AOS and suggest that other regulators of these Rho GTPase proteins might be good candidates in the quest to define the genetic spectrum of this genetically heterogeneous condition.
Copyright © 2011 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Adams F.H., Oliver C.P. Hereditary deformities in man due to arrested development. J. Hered. 1945;36:3–7.
-
- Whitley C.B., Gorlin R.J. Adams-Oliver syndrome revisited. Am. J. Med. Genet. 1991;40:319–326. - PubMed
-
- Snape K.M., Ruddy D., Zenker M., Wuyts W., Whiteford M., Johnson D., Lam W., Trembath R.C. The spectra of clinical phenotypes in aplasia cutis congenita and terminal transverse limb defects. Am. J. Med. Genet. A. 2009;149A:1860–1881. - PubMed
-
- Papadopoulou E., Sifakis S., Raissaki M., Germanakis I., Kalmanti M. Antenatal and postnatal evidence of periventricular leukomalacia as a further indication of vascular disruption in Adams-Oliver syndrome. Am. J. Med. Genet. A. 2008;146A:2545–2550. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
